



Biotech Depot 2008 - 500 Beiträge pro Seite
eröffnet am 03.08.08 17:44:52 von
neuester Beitrag 11.01.09 16:18:50 von
neuester Beitrag 11.01.09 16:18:50 von
Beiträge: 126
ID: 1.143.292
ID: 1.143.292
Aufrufe heute: 1
Gesamt: 46.195
Gesamt: 46.195
Aktive User: 0
Top-Diskussionen
| Titel | letzter Beitrag | Aufrufe |
|---|---|---|
| gestern 23:11 | 499 | |
| heute 00:52 | 263 | |
| gestern 21:36 | 158 | |
| gestern 23:28 | 147 | |
| vor 48 Minuten | 143 | |
| heute 00:16 | 139 | |
| vor 1 Stunde | 131 | |
| vor 1 Stunde | 130 |
Meistdiskutierte Wertpapiere
| Platz | vorher | Wertpapier | Kurs | Perf. % | Anzahl | ||
|---|---|---|---|---|---|---|---|
| 1. | 1. | 18.668,00 | -0,69 | 205 | |||
| 2. | 3. | 0,7757 | +15,60 | 93 | |||
| 3. | 2. | 2,6950 | -41,09 | 89 | |||
| 4. | 4. | 176,75 | -1,39 | 66 | |||
| 5. | 6. | 23,740 | +24,95 | 66 | |||
| 6. | 5. | 0,1930 | -4,93 | 59 | |||
| 7. | 8. | 1.139,01 | +6,98 | 42 | |||
| 8. | 7. | 2.358,14 | -0,13 | 39 |
Hallo!
Soll ich es nochmal wagen nach dem GPC-Desaster im letzten Jahr (Thread: Biotech Depot 2007)? 2008 ist schon halb rum... daher nun nach dem katastrophalen Biotech-Depot 2007 sehr verspätet nun wieder das neue Depot der reinen Biotechs 2008! - Neues Jahr neues Glück?
- Neues Jahr neues Glück? 
Die einzige Lehre, die ich aus dem letzten Jahr ziehen kann: nie wieder eine so große Position auf einen Wert konzentrieren, speziell wenn es sich dabei um ein Ein-Produkt-Unternehmen handelt. Daher orientiere ich mich seitdem an den mittleren, bereits etwas teureren Biotechs, die entweder bereits profitabel sind oder eine breite Pipeline besitzen, viel Cash, hohe Investitionen in die Entwicklung, Produkte möglichst weit in der klinischen Entwicklung, potentielle Blockbuster und namhafte Partner. Zudem soll das Depot eine höhere Streuung erhalten. Der zurückliegende Monat ist ausnahmsweise mal sehr positiv gelaufen, so dass ich es wage, einen entsprechenden Thread zu meinen Biotech-Werten zu erstellen.
So sieht es aktuell im Depot aus:
24,3% Genmab http://finance.yahoo.com/q?s=GEN.CO
15,4% Regeneron http://finance.yahoo.com/q?s=REGN
9,5% Onyx http://finance.yahoo.com/q?s=ONXX
7,2% OSI Pharma http://finance.yahoo.com/q?s=OSIP
6,3% (KO) Intercell http://finance.yahoo.com/q?s=IJE.F
5,4% Vertex http://finance.yahoo.com/q?s=VRTX
5,3% Cubist http://finance.yahoo.com/q?s=CBST
5,1% Imclone http://finance.yahoo.com/q?s=IMCL
5,1% Evotec http://finance.yahoo.com/q?s=EVT.DE
4,8% Isis http://finance.yahoo.com/q?s=ISIS
4,0% Arena http://finance.yahoo.com/q?s=ARNA
3,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
2,7% Micromet http://finance.yahoo.com/q?s=MITI
1,9% Supergen http://finance.yahoo.com/q?s=SUPG
auf meiner Watchliste stehen INCY, RIGL und POZN. In der zweiten Reihe SGEN, Basilea, EXEL und MNTA.
Aus dem alten Depot ist nur noch Genmab und Arena übrig geblieben... GPC hat sich nahezu aufgelöst, MOR, POZN und CRME habe ich verkauft, während Speedel kürzlich übernommen wurde.
Ich hoffe, dies wird ein besseres Jahr! Der NBI steht momentan gar nicht schlecht... kann sich aber auch schnell wieder ändern.

mfg ipollit
Soll ich es nochmal wagen nach dem GPC-Desaster im letzten Jahr (Thread: Biotech Depot 2007)? 2008 ist schon halb rum... daher nun nach dem katastrophalen Biotech-Depot 2007 sehr verspätet nun wieder das neue Depot der reinen Biotechs 2008!
 - Neues Jahr neues Glück?
- Neues Jahr neues Glück? 
Die einzige Lehre, die ich aus dem letzten Jahr ziehen kann: nie wieder eine so große Position auf einen Wert konzentrieren, speziell wenn es sich dabei um ein Ein-Produkt-Unternehmen handelt. Daher orientiere ich mich seitdem an den mittleren, bereits etwas teureren Biotechs, die entweder bereits profitabel sind oder eine breite Pipeline besitzen, viel Cash, hohe Investitionen in die Entwicklung, Produkte möglichst weit in der klinischen Entwicklung, potentielle Blockbuster und namhafte Partner. Zudem soll das Depot eine höhere Streuung erhalten. Der zurückliegende Monat ist ausnahmsweise mal sehr positiv gelaufen, so dass ich es wage, einen entsprechenden Thread zu meinen Biotech-Werten zu erstellen.
So sieht es aktuell im Depot aus:
24,3% Genmab http://finance.yahoo.com/q?s=GEN.CO
15,4% Regeneron http://finance.yahoo.com/q?s=REGN
9,5% Onyx http://finance.yahoo.com/q?s=ONXX
7,2% OSI Pharma http://finance.yahoo.com/q?s=OSIP
6,3% (KO) Intercell http://finance.yahoo.com/q?s=IJE.F
5,4% Vertex http://finance.yahoo.com/q?s=VRTX
5,3% Cubist http://finance.yahoo.com/q?s=CBST
5,1% Imclone http://finance.yahoo.com/q?s=IMCL
5,1% Evotec http://finance.yahoo.com/q?s=EVT.DE
4,8% Isis http://finance.yahoo.com/q?s=ISIS
4,0% Arena http://finance.yahoo.com/q?s=ARNA
3,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
2,7% Micromet http://finance.yahoo.com/q?s=MITI
1,9% Supergen http://finance.yahoo.com/q?s=SUPG
auf meiner Watchliste stehen INCY, RIGL und POZN. In der zweiten Reihe SGEN, Basilea, EXEL und MNTA.
Aus dem alten Depot ist nur noch Genmab und Arena übrig geblieben... GPC hat sich nahezu aufgelöst, MOR, POZN und CRME habe ich verkauft, während Speedel kürzlich übernommen wurde.
Ich hoffe, dies wird ein besseres Jahr! Der NBI steht momentan gar nicht schlecht... kann sich aber auch schnell wieder ändern.
mfg ipollit

hier die zugehörigen Charts...
Genmab

Regeneron

Onyx

OSI Pharma

(KO) Intercell

Vertex

Cubist

Imclone

Evotec

Isis

Arena

ViroPharma

Micromet

Supergen

Genmab
Regeneron
Onyx
OSI Pharma
(KO) Intercell
Vertex
Cubist
Imclone
Evotec
Isis
Arena
ViroPharma
Micromet
Supergen
und die Watch-Liste...
Incyte

Rigel

Pozen

mfg ipollit
Incyte
Rigel
Pozen
mfg ipollit
Genmab
http://www.genmab.com/
Marktkapitalisierung: 3,05 Mrd USD
Genmab ging aus Medarex hervor. Neben der AK-Technologie besitzt Genmab auch UniBody-AKs, die nur einen von zwei Armen besitzen, wodurch sich z.B. IgG4-AKs stabilisieren lassen. Außerdem besitzt Produktionsanlagen für die kommerzielle Herstellung von AKs.
die aktuelle Pipeline:
der wichtigste Kandidat ist Ofatumumab (Humax-CD20), der in einer 2 Mrd USD Kooperation zusammen mit GSK entwickelt wird. Ofatumumab ist eine humane Version von Rituxan. Die Indikationen sind demnach Blutkrebsarten wie CLL oder NHL, sowie Rheumatische Arthritis. Angepeilte peak sales sind bis zu mehrere Mrd USD.
Zalutumumab ist eine humane Version des Antikörpers Erbitux.

29.05.2008 | 11:45 Uhr
Genmab meldet Ergebnisse für das erste Quartal 2008
Kopenhagen (ots/PRNewswire) - - Zusammenfassung: Genmab meldet Ergebnisse für die ersten drei Monate des Jahres 2008
Genmab A/S (OMX: GEN) gab heute die Ergebnisse für das Quartal zum 31. März 2008 bekannt. Für diesen Zeitraum meldete Genmab die folgenden Ergebnisse:
Der Umsatz von Genmab lag für das erste Quartal 2008 bei 167 Mio. DKK (ca. 36 Mio. USD). Während des Vergleichszeitraums im Jahr 2007 erwirtschaftete Genmab einen Umsatz von 80 Mio. DKK (ca. 17 Mio. USD).
Der operative Verlust beläuft sich auf 197 Mio. DKK (ca. 42 Mio. USD). Verglichen hiermit lag der operative Verlust für den gleichen Zeitraum des Jahres 2007 bei 106 Mio. DKK (ca. 22 Mio. USD).
Im Nettofinanzertrag für das erste Quartal 2008 ist ein Nettoverlust von 14 Mio. DKK (ca. 3 Mio. USD) enthalten, verglichen mit einem Nettoertrag von 29 Mio. DKK (ca. 6 Mio. USD) im gleichen Zeitraum des Jahres 2007. Das Nettofinanzergebnis setzt sich einerseits zusammen aus den positiven Zinserträgen aus unseren Portfolios börsengängiger Wertpapiere, andererseits spiegeln sich darin die nicht realisierten Devisenverluste wider, die als Folge der anhaltenden Schwächung des US-Dollars gegenüber der Dänischen Krone im ersten Quartal 2008 entstanden sind.
Der Nettoverlust beläuft sich auf 210 Mio. DKK (ca. 45 Mio. USD), gegenüber einem Nettoverlust von 77 Mio. DKK (ca. 16 Mio. USD) im gleichen Zeitraum des Jahres 2007. Der Nettoverlust je Aktie belief sich im ersten Quartal 2008 auf 4,73 DKK (ca. 1,00 USD), verglichen mit 1,81 DKK (ca. 0,38 USD) im ersten Quartal 2007.
Genmab verzeichnete zum Ende des ersten Quartals einen Barmittelbestand von 2,4 Mrd. DKK (ca. 503 Mio. USD). Dies entspricht einem Rückgang um 1,3 Mrd. DKK (ca. 280 Mio. USD) gegenüber dem Bestand zum Ende des Jahres 2007. Der Rückgang ist dabei hauptsächlich auf den Kauf einer Produktionsanlage für 1,2 Mrd. DKK (ca. 240 Mio. USD zum Zeitpunkt des Kaufs) im März 2008 zurückzuführen.
Höhepunkte: Im Verlauf des ersten Quartals 2008 konnte Genmab etliche geschäftliche und wissenschaftliche Meilensteine erreichen, u.a.:
Im März erwarb Genmab eine Antikörper-Produktionsanlage von PDL BioPharma zum Preis von 1,2 Mrd. DKK (240 Mio. USD zum Zeitpunkt des Kaufs).
Im Januar meldete Genmab die Entwicklung eines neuen vorklinischen Produkts namens HuMax-CD32b. Der Antikörper könnte möglicherweise über therapeutisches Potenzial bei der Behandlung chronischer lymphatischer B-Zellen-Leukämie, kleinzelliger lymphatischer Lymphome, Burkitt-Lymphome, follikulärer Lymphome und diffuser grosszelliger B-Zellen-Lymphome verfügen.
Im Rahmen der Zusammenarbeit mit GlaxoSmithKline erreichten wir im Januar den dritten Meilenstein, als der erste Patient im Rahmen des Phase-III-RA-Programms behandelt wurde und wir eine Zahlung über 87 Mio. DKK (ca. 18 Mio. USD) erhielten.
Ausblick: Genmab behält seine Finanzprognose für das Jahr bei und erwartet weiterhin einen operativen Verlust von 900 Mio. bis 1 Mrd. DKK (ca. 187 bis 208 Mio. USD)sowie einen Nettoverlust im Bereich von 800 bis 900 Mio. DKK (ca. 167 bis 187 Mio. USD). Es wird erwartet, dass der Umsatz für 2008 ca. 1,0 Mrd. DKK (ca. 208 Mio. USD) betragen wird.
Zum Stichtag 31. Dezember 2007 verfügte Genmab über Barmittel und befristete börsengängige Wertpapiere in Höhe von 3,7 Mrd. DKK. Für 2008 erwarten wir, dass sich durch unsere Geschäftstätigkeiten sowie durch den Kauf der Produktionsanlage in Minnesota für 1,2 Mrd. DKK eine Barliquidität von 1,7 bis 1,8 Mrd. DKK (ca. 360 Mio. bis 382 Mio. USD) zum Jahresende ergibt.
mfg ipollit
http://www.genmab.com/
Marktkapitalisierung: 3,05 Mrd USD
Genmab ging aus Medarex hervor. Neben der AK-Technologie besitzt Genmab auch UniBody-AKs, die nur einen von zwei Armen besitzen, wodurch sich z.B. IgG4-AKs stabilisieren lassen. Außerdem besitzt Produktionsanlagen für die kommerzielle Herstellung von AKs.
die aktuelle Pipeline:
der wichtigste Kandidat ist Ofatumumab (Humax-CD20), der in einer 2 Mrd USD Kooperation zusammen mit GSK entwickelt wird. Ofatumumab ist eine humane Version von Rituxan. Die Indikationen sind demnach Blutkrebsarten wie CLL oder NHL, sowie Rheumatische Arthritis. Angepeilte peak sales sind bis zu mehrere Mrd USD.
Zalutumumab ist eine humane Version des Antikörpers Erbitux.
29.05.2008 | 11:45 Uhr
Genmab meldet Ergebnisse für das erste Quartal 2008
Kopenhagen (ots/PRNewswire) - - Zusammenfassung: Genmab meldet Ergebnisse für die ersten drei Monate des Jahres 2008
Genmab A/S (OMX: GEN) gab heute die Ergebnisse für das Quartal zum 31. März 2008 bekannt. Für diesen Zeitraum meldete Genmab die folgenden Ergebnisse:
Der Umsatz von Genmab lag für das erste Quartal 2008 bei 167 Mio. DKK (ca. 36 Mio. USD). Während des Vergleichszeitraums im Jahr 2007 erwirtschaftete Genmab einen Umsatz von 80 Mio. DKK (ca. 17 Mio. USD).
Der operative Verlust beläuft sich auf 197 Mio. DKK (ca. 42 Mio. USD). Verglichen hiermit lag der operative Verlust für den gleichen Zeitraum des Jahres 2007 bei 106 Mio. DKK (ca. 22 Mio. USD).
Im Nettofinanzertrag für das erste Quartal 2008 ist ein Nettoverlust von 14 Mio. DKK (ca. 3 Mio. USD) enthalten, verglichen mit einem Nettoertrag von 29 Mio. DKK (ca. 6 Mio. USD) im gleichen Zeitraum des Jahres 2007. Das Nettofinanzergebnis setzt sich einerseits zusammen aus den positiven Zinserträgen aus unseren Portfolios börsengängiger Wertpapiere, andererseits spiegeln sich darin die nicht realisierten Devisenverluste wider, die als Folge der anhaltenden Schwächung des US-Dollars gegenüber der Dänischen Krone im ersten Quartal 2008 entstanden sind.
Der Nettoverlust beläuft sich auf 210 Mio. DKK (ca. 45 Mio. USD), gegenüber einem Nettoverlust von 77 Mio. DKK (ca. 16 Mio. USD) im gleichen Zeitraum des Jahres 2007. Der Nettoverlust je Aktie belief sich im ersten Quartal 2008 auf 4,73 DKK (ca. 1,00 USD), verglichen mit 1,81 DKK (ca. 0,38 USD) im ersten Quartal 2007.
Genmab verzeichnete zum Ende des ersten Quartals einen Barmittelbestand von 2,4 Mrd. DKK (ca. 503 Mio. USD). Dies entspricht einem Rückgang um 1,3 Mrd. DKK (ca. 280 Mio. USD) gegenüber dem Bestand zum Ende des Jahres 2007. Der Rückgang ist dabei hauptsächlich auf den Kauf einer Produktionsanlage für 1,2 Mrd. DKK (ca. 240 Mio. USD zum Zeitpunkt des Kaufs) im März 2008 zurückzuführen.
Höhepunkte: Im Verlauf des ersten Quartals 2008 konnte Genmab etliche geschäftliche und wissenschaftliche Meilensteine erreichen, u.a.:
Im März erwarb Genmab eine Antikörper-Produktionsanlage von PDL BioPharma zum Preis von 1,2 Mrd. DKK (240 Mio. USD zum Zeitpunkt des Kaufs).
Im Januar meldete Genmab die Entwicklung eines neuen vorklinischen Produkts namens HuMax-CD32b. Der Antikörper könnte möglicherweise über therapeutisches Potenzial bei der Behandlung chronischer lymphatischer B-Zellen-Leukämie, kleinzelliger lymphatischer Lymphome, Burkitt-Lymphome, follikulärer Lymphome und diffuser grosszelliger B-Zellen-Lymphome verfügen.
Im Rahmen der Zusammenarbeit mit GlaxoSmithKline erreichten wir im Januar den dritten Meilenstein, als der erste Patient im Rahmen des Phase-III-RA-Programms behandelt wurde und wir eine Zahlung über 87 Mio. DKK (ca. 18 Mio. USD) erhielten.
Ausblick: Genmab behält seine Finanzprognose für das Jahr bei und erwartet weiterhin einen operativen Verlust von 900 Mio. bis 1 Mrd. DKK (ca. 187 bis 208 Mio. USD)sowie einen Nettoverlust im Bereich von 800 bis 900 Mio. DKK (ca. 167 bis 187 Mio. USD). Es wird erwartet, dass der Umsatz für 2008 ca. 1,0 Mrd. DKK (ca. 208 Mio. USD) betragen wird.
Zum Stichtag 31. Dezember 2007 verfügte Genmab über Barmittel und befristete börsengängige Wertpapiere in Höhe von 3,7 Mrd. DKK. Für 2008 erwarten wir, dass sich durch unsere Geschäftstätigkeiten sowie durch den Kauf der Produktionsanlage in Minnesota für 1,2 Mrd. DKK eine Barliquidität von 1,7 bis 1,8 Mrd. DKK (ca. 360 Mio. bis 382 Mio. USD) zum Jahresende ergibt.
mfg ipollit
Was aber der Evotec Schrott darin zu suchen hat, erschließt sich mir net. Evotec wird sterben wie GPC.
Ansonsten auf alle Fälle Adolor Corp (ADLR)..
Und Alnylam (ALNY) gehört eigentlich auch rein! Jedenfalls wenn Isis dabei ist.
Herz5!
Ansonsten auf alle Fälle Adolor Corp (ADLR)..
Und Alnylam (ALNY) gehört eigentlich auch rein! Jedenfalls wenn Isis dabei ist.
Herz5!
Trading Spotlight
Antwort auf Beitrag Nr.: 34.639.011 von ipollit am 03.08.08 19:38:58aktuell positive PIII-Daten von HuMax-CD20 bei CLL... Ziel war eine Ansprechrate von 25%, erreicht wurde eine deutlich höhere. Damit wäre eine Zulassung für diese erste Indikation im nächsten Jahr möglich.
***********
01.08.2008 | 14:30 Uhr
Genmab und GlaxoSmithKline melden positive Top-Line-Ergebnisse der Pivotstudie mit Ofatumumab zur Behandlung von chronischer lymphatischer Leukämie
Kopenhagen (ots/PRNewswire) - - Zusammenfassung: Pivotstudie der Phase III für Ofatumumab bei refraktärer CLL erreicht primären Endpunkt
Genmab A/S (OMX: GEN) und GlaxoSmithKline (LSE und NYSE: GSK) meldeten heute, dass eine vorläufige Analyse der Pivotstudie der Phase III positive Ergebnisse der wesentlichen Eckpunkte (Top-Line-Ergebnisse) ergeben hätte. Im Rahmen der Studie wurde der Einsatz von Ofatumumab (HuMax-CD20(R)) bei der Behandlung von zwei Patientengruppen mit chronischer lymphatischer Leukämie (CLL) bewertet, für die es bisher kaum Therapieoptionen gibt. Zum Zeitpunkt der vorläufigen Analyse wurde bei beiden Populationen der primäre Endpunkt der Studie erreicht; auch die Ergebnisse der sekundären Endpunkte unterstützen den primären Endpunkt.
In dieser vorläufigen Analyse wurde die Aktivität von Ofatumumab bei 154 Patienten bewertet; bei 138 dieser Patienten mit refraktärer CLL waren die Ergebnisse tatsächlich auswertbar. Etwa die Hälfte der an der Studie teilnehmenden Patienten (59) waren sowohl für Fludarabin als auch für Alemtuzumab refraktär. Im Rahmen der Analyse wurde auch eine zweite Gruppe (79) untersucht, die Fludarabin-refraktär waren und aufgrund grosser Tumore in den Lymphknoten als ungeeignete Kandidaten für Alemtuzumab angesehen wurden. In der Patientengruppe, die sowohl Fludarabin- als auch Alemtuzumab-refraktär waren, wurde eine objektive Ansprechrate von 51 % (p < 0.0001), bestehend aus 30 partiellen Reaktionen (PR), erreicht. In der Fludarabin-refraktären und für Alemtuzumab ungeeigneten Patientengruppe wurde eine objektive Ansprechrate von 44 % (p < 0.0001), darunter 1 vollständige Reaktion (CR) und 34 PR, festgestellt. Dass diese objektiven Ansprechraten, wie festgestellt erreicht wurden, geht aus Bewertungen eines unabhängigen Komitees hervor und unterliegt der Prüfung und Bestätigung durch die Zulassungsbehörden.
Ofatumumab wurde von CLL-Patienten in der Studie im Allgemeinen gut vertragen. Die am häufigsten festgestellten Nebenwirkungen (mit einer Häufigkeit von mehr als 15 %) waren: Pyrexie, Durchfall, Müdigkeit, Husten, Neutropenie, Anämie und Pneumonie. Es traten keine unerwarteten Sicherheitsrisiken auf. Bei keinem der 14 auf humane Antihuman-Antikörper (HAHA) getesteten Patienten wurden diese 12 Monate später nachgewiesen.
Eine Vorbesprechung zur Biologics License Application (BLA) wurde bei der FDA beantragt, bei der diese Ergebnisse erörtert werden sollen, so dass möglicherweise schon 2008 eine BLA eingereicht werden kann. Auch eine Einreichung bei den Zulassungsbehörden der EU kann möglicherweise im selben Zeitrahmen realisiert werden. Die vollständigen Daten werden zu gegebener Zeit im Rahmen einer wissenschaftlichen Fachkonferenz vorgestellt werden.
"Wir sind sehr erfreut, positive Ergebnisse bezüglich der Behandlung der an dieser Studie teilnehmenden CLL-Patienten bekannt geben zu können", so Dr. Lisa N. Drakeman, Chief Executive Officer von Genmab. "Auch für Genmab ist dies ein bedeutender Schritt. Wir stehen nun kurz vor der Einreichung des ersten Antrags auf Marktzulassung für einen Genmab-Antikörper und freuen uns darauf, bei der Einreichung der Unterlagen mit GSK zusammenzuarbeiten."
"Diese sehr vielversprechenden Ergebnisse weisen darauf hin, dass Ofatumumab das Potenzial hat, CLL-Patienten mit einer äusserst refraktären Erkrankung und eingeschränkten Behandlungsmöglichkeiten Vorteile zu bieten", sagte Kathy Rouan, Vice President und Leiterin der Medizinischen Entwicklung bei GSK. "GSK und Genmab arbeiten gemeinsam an einem umfassenden Entwicklungsprogramm für CLL und für Nicht-Hodgkins-Lymphome (NHL). Wir hoffen, dass diese sowohl für Patienten als auch für deren Ärzte einen bedeutenden Beitrag zur Behandlung dieser bösartigen hämatologischen Tumoren leisten werden."
Ofatumumab ist ein in der Entwicklung befindlicher, vollständig humaner, monoklonaler Antikörper der nächsten Generation, der sich an das kleine Schleifen-Epitop (spezifische Antikörper-Bindungsposition) auf dem CD20-Rezeptor auf der Oberfläche von B-Zellen bindet. Im Rahmen einer Vereinbarung zwischen Genmab und GlaxoSmithKline zur gemeinsamen Entwicklung und Vermarktung von Ofatumumab wird dieser Wirkstoff derzeit zur Behandlung der CLL, des follikulären Nicht-Hodgkin-Lymphoms, des diffusen grosszelligen B-Zellen-Lymphoms, der rheumatoiden Arthritis und der rezidivierenden, remittierenden multiplen Sklerose entwickelt. Ofatumumab ist bisher noch in keinem Land zugelassen.
mfg ipollit
***********
01.08.2008 | 14:30 Uhr
Genmab und GlaxoSmithKline melden positive Top-Line-Ergebnisse der Pivotstudie mit Ofatumumab zur Behandlung von chronischer lymphatischer Leukämie
Kopenhagen (ots/PRNewswire) - - Zusammenfassung: Pivotstudie der Phase III für Ofatumumab bei refraktärer CLL erreicht primären Endpunkt
Genmab A/S (OMX: GEN) und GlaxoSmithKline (LSE und NYSE: GSK) meldeten heute, dass eine vorläufige Analyse der Pivotstudie der Phase III positive Ergebnisse der wesentlichen Eckpunkte (Top-Line-Ergebnisse) ergeben hätte. Im Rahmen der Studie wurde der Einsatz von Ofatumumab (HuMax-CD20(R)) bei der Behandlung von zwei Patientengruppen mit chronischer lymphatischer Leukämie (CLL) bewertet, für die es bisher kaum Therapieoptionen gibt. Zum Zeitpunkt der vorläufigen Analyse wurde bei beiden Populationen der primäre Endpunkt der Studie erreicht; auch die Ergebnisse der sekundären Endpunkte unterstützen den primären Endpunkt.
In dieser vorläufigen Analyse wurde die Aktivität von Ofatumumab bei 154 Patienten bewertet; bei 138 dieser Patienten mit refraktärer CLL waren die Ergebnisse tatsächlich auswertbar. Etwa die Hälfte der an der Studie teilnehmenden Patienten (59) waren sowohl für Fludarabin als auch für Alemtuzumab refraktär. Im Rahmen der Analyse wurde auch eine zweite Gruppe (79) untersucht, die Fludarabin-refraktär waren und aufgrund grosser Tumore in den Lymphknoten als ungeeignete Kandidaten für Alemtuzumab angesehen wurden. In der Patientengruppe, die sowohl Fludarabin- als auch Alemtuzumab-refraktär waren, wurde eine objektive Ansprechrate von 51 % (p < 0.0001), bestehend aus 30 partiellen Reaktionen (PR), erreicht. In der Fludarabin-refraktären und für Alemtuzumab ungeeigneten Patientengruppe wurde eine objektive Ansprechrate von 44 % (p < 0.0001), darunter 1 vollständige Reaktion (CR) und 34 PR, festgestellt. Dass diese objektiven Ansprechraten, wie festgestellt erreicht wurden, geht aus Bewertungen eines unabhängigen Komitees hervor und unterliegt der Prüfung und Bestätigung durch die Zulassungsbehörden.
Ofatumumab wurde von CLL-Patienten in der Studie im Allgemeinen gut vertragen. Die am häufigsten festgestellten Nebenwirkungen (mit einer Häufigkeit von mehr als 15 %) waren: Pyrexie, Durchfall, Müdigkeit, Husten, Neutropenie, Anämie und Pneumonie. Es traten keine unerwarteten Sicherheitsrisiken auf. Bei keinem der 14 auf humane Antihuman-Antikörper (HAHA) getesteten Patienten wurden diese 12 Monate später nachgewiesen.
Eine Vorbesprechung zur Biologics License Application (BLA) wurde bei der FDA beantragt, bei der diese Ergebnisse erörtert werden sollen, so dass möglicherweise schon 2008 eine BLA eingereicht werden kann. Auch eine Einreichung bei den Zulassungsbehörden der EU kann möglicherweise im selben Zeitrahmen realisiert werden. Die vollständigen Daten werden zu gegebener Zeit im Rahmen einer wissenschaftlichen Fachkonferenz vorgestellt werden.
"Wir sind sehr erfreut, positive Ergebnisse bezüglich der Behandlung der an dieser Studie teilnehmenden CLL-Patienten bekannt geben zu können", so Dr. Lisa N. Drakeman, Chief Executive Officer von Genmab. "Auch für Genmab ist dies ein bedeutender Schritt. Wir stehen nun kurz vor der Einreichung des ersten Antrags auf Marktzulassung für einen Genmab-Antikörper und freuen uns darauf, bei der Einreichung der Unterlagen mit GSK zusammenzuarbeiten."
"Diese sehr vielversprechenden Ergebnisse weisen darauf hin, dass Ofatumumab das Potenzial hat, CLL-Patienten mit einer äusserst refraktären Erkrankung und eingeschränkten Behandlungsmöglichkeiten Vorteile zu bieten", sagte Kathy Rouan, Vice President und Leiterin der Medizinischen Entwicklung bei GSK. "GSK und Genmab arbeiten gemeinsam an einem umfassenden Entwicklungsprogramm für CLL und für Nicht-Hodgkins-Lymphome (NHL). Wir hoffen, dass diese sowohl für Patienten als auch für deren Ärzte einen bedeutenden Beitrag zur Behandlung dieser bösartigen hämatologischen Tumoren leisten werden."
Ofatumumab ist ein in der Entwicklung befindlicher, vollständig humaner, monoklonaler Antikörper der nächsten Generation, der sich an das kleine Schleifen-Epitop (spezifische Antikörper-Bindungsposition) auf dem CD20-Rezeptor auf der Oberfläche von B-Zellen bindet. Im Rahmen einer Vereinbarung zwischen Genmab und GlaxoSmithKline zur gemeinsamen Entwicklung und Vermarktung von Ofatumumab wird dieser Wirkstoff derzeit zur Behandlung der CLL, des follikulären Nicht-Hodgkin-Lymphoms, des diffusen grosszelligen B-Zellen-Lymphoms, der rheumatoiden Arthritis und der rezidivierenden, remittierenden multiplen Sklerose entwickelt. Ofatumumab ist bisher noch in keinem Land zugelassen.
mfg ipollit
Antwort auf Beitrag Nr.: 34.639.011 von ipollit am 03.08.08 19:38:5801.07.2008 | 07:44 Uhr
Genmab erreicht Meilenstein in der Ofatumumab-Zusammenarbeit
Kopenhagen (ots/PRNewswire) - - Zusammenfassung: Genmab hat einen Meilenstein in seiner Ofatumumab-Zusammenarbeit mit GlaxoSmithKline erreicht.
Genmab A/S (OMX: GEN) hat heute bekannt gegeben, dass es im Rahmen seiner Zusammenarbeit mit GlaxoSmithKline (GSK) einen Entwicklungs-Meilenstein für Ofatumumab (HuMax-CD20(R)) erreicht hat. Eine Meilenstein-Zahlung von schätzungsweise 29 Millionen DKK (schätzungsweise 6 Millionen USD) wurde durch die Behandlung des ersten Patienten ausgelöst, der an der Phase II-Studie von Ofatumumab zur Behandlung von schubförmig remittierender Multipler Sklerose (RRMS) teilgenommen hat.
"Unsere Zusammenarbeit mit GSK macht weiter Fortschritte, da beide Unternehmen darauf hinarbeiten, neue Behandlungsmöglichkeiten für Patienten anzubieten", erklärte Lisa N. Drakeman, Ph.D., Geschäftsführerin von Genmab. "Wir freuen uns, in der ersten Studie von Ofatumumab bei RRMS, einer unberechenbaren und starke Einschränkungen mit sich bringenden Erkrankung, mit der Behandlung von Patienten zu beginnen."
Ofatumumab ist ein in der Entwicklung befindlicher, vollständig menschlicher, monoklonaler Antikörper der nächsten Generation, der sich an ein ausgeprägtes, kleines, schleifenförmiges Epitop auf dem CD20-Rezeptor auf der Oberfläche von B-Zellen bindet. Dieses Epitop unterscheidet sich von den anderen Anti-CD20-Antikörpern, die zurzeit verfügbar sind oder sich in der Entwicklung befinden. Ofatumumab wird im Rahmen einer gemeinsamen Entwicklungs- und Vermarktungsvereinbarung zwischen Genmab und GSK entwickelt.
mfg ipollit
Genmab erreicht Meilenstein in der Ofatumumab-Zusammenarbeit
Kopenhagen (ots/PRNewswire) - - Zusammenfassung: Genmab hat einen Meilenstein in seiner Ofatumumab-Zusammenarbeit mit GlaxoSmithKline erreicht.
Genmab A/S (OMX: GEN) hat heute bekannt gegeben, dass es im Rahmen seiner Zusammenarbeit mit GlaxoSmithKline (GSK) einen Entwicklungs-Meilenstein für Ofatumumab (HuMax-CD20(R)) erreicht hat. Eine Meilenstein-Zahlung von schätzungsweise 29 Millionen DKK (schätzungsweise 6 Millionen USD) wurde durch die Behandlung des ersten Patienten ausgelöst, der an der Phase II-Studie von Ofatumumab zur Behandlung von schubförmig remittierender Multipler Sklerose (RRMS) teilgenommen hat.
"Unsere Zusammenarbeit mit GSK macht weiter Fortschritte, da beide Unternehmen darauf hinarbeiten, neue Behandlungsmöglichkeiten für Patienten anzubieten", erklärte Lisa N. Drakeman, Ph.D., Geschäftsführerin von Genmab. "Wir freuen uns, in der ersten Studie von Ofatumumab bei RRMS, einer unberechenbaren und starke Einschränkungen mit sich bringenden Erkrankung, mit der Behandlung von Patienten zu beginnen."
Ofatumumab ist ein in der Entwicklung befindlicher, vollständig menschlicher, monoklonaler Antikörper der nächsten Generation, der sich an ein ausgeprägtes, kleines, schleifenförmiges Epitop auf dem CD20-Rezeptor auf der Oberfläche von B-Zellen bindet. Dieses Epitop unterscheidet sich von den anderen Anti-CD20-Antikörpern, die zurzeit verfügbar sind oder sich in der Entwicklung befinden. Ofatumumab wird im Rahmen einer gemeinsamen Entwicklungs- und Vermarktungsvereinbarung zwischen Genmab und GSK entwickelt.
mfg ipollit
Antwort auf Beitrag Nr.: 34.639.023 von -Salem- am 03.08.08 19:45:12Richtig, Alnylam habe ich auch auf meiner Liste gehabt, war mir dann aber doch zu teuer (1,44 Mrd USD MK!!!). Angeblich sollen sie zwar das Maß bezüglich RNAi sein, aber ob diese Technologie wirklich die Wunderwaffe ist, muss sich noch zeigen. Über 1 Mrd USD MK und so gut wie nichts in der klinischen Pipeline riecht mir etwas zu sehr nach Hype.
Statt ADLR würde ich eher PGNX nehmen, oder?
Evotec? Ist für mich im Moment der einzige deutsche Biotech, der interessant ist. Halte ich für äußerst vielversprechend mit einer spannenden CNS-Pipeline: u.a. Schlafmittel, Raucherentwöhnungsmittel und Alzheimer-Medikament... alles potentielle Blockbuster, teilweise schon mit POC. Und ausreichend Cash für die nächste Zeit.
mfg ipollit
Statt ADLR würde ich eher PGNX nehmen, oder?
Evotec? Ist für mich im Moment der einzige deutsche Biotech, der interessant ist. Halte ich für äußerst vielversprechend mit einer spannenden CNS-Pipeline: u.a. Schlafmittel, Raucherentwöhnungsmittel und Alzheimer-Medikament... alles potentielle Blockbuster, teilweise schon mit POC. Und ausreichend Cash für die nächste Zeit.
mfg ipollit
Antwort auf Beitrag Nr.: 34.639.011 von ipollit am 03.08.08 19:38:58hier ein Überlick über potentiell anstehende Events in der nächsten Zeit: Die CLL PIII-Daten sind bereits bekannt.
•Ofatumumab: Phase III data for the CLL indication (expected in August 2008).
•Ofatumumab: Phase III data for the FL indication (expected in September/October 2008).
•Zalutumumab: Interim phase-III data for the head and neck cancer indication (expected in Q4 2008).
•Ofatumumab: Approval of the CLL indication (expected in Q4, 2008).
•Zalutumumab: Partnership agreement.
•Unibody: Technology update
•Unibody: Partnership agreement.
•Zanolimumab: Phase III data for the CTCL indication (expected in 2009, 2H).
mfg ipollit
•Ofatumumab: Phase III data for the CLL indication (expected in August 2008).
•Ofatumumab: Phase III data for the FL indication (expected in September/October 2008).
•Zalutumumab: Interim phase-III data for the head and neck cancer indication (expected in Q4 2008).
•Ofatumumab: Approval of the CLL indication (expected in Q4, 2008).
•Zalutumumab: Partnership agreement.
•Unibody: Technology update
•Unibody: Partnership agreement.
•Zanolimumab: Phase III data for the CTCL indication (expected in 2009, 2H).
mfg ipollit
Und warum fehlt Actelion?
Antwort auf Beitrag Nr.: 34.639.625 von Matze900 am 03.08.08 23:32:19Actelion... Weil ich nicht beurteilen konnte, welchen Einfluss Letairis und Thelin auf die Tracleer-Umsätze haben werden. Actelion ist von Tracleer noch ziemlich abhängig. Die MK ist mit 6,7 Mrd USD auch nicht so niedrig, dass es geradezu ein Schnäppchen ist. Werde ich mir aber nochmal genauer ansehen. Sollte Tracleer stabil wachsen, dann ist Actelion vor dem Hintergrund von Almorexant allerdings eine Investition wert.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 34.642.261 von ipollit am 04.08.08 13:21:45
Published: 07:00 04.08.2008 GMT+2 /HUGIN /Source: Actelion Pharmaceuticals Ltd /SWX: ATLN /ISIN: CH0010532478
Tracleer® (bosentan) receives EU approval for treatment of patients with mildly symptomatic Pulmonary Arterial Hypertension
ALLSCHWIL/BASEL, SWITZERLAND - 04 August 2008 - Actelion Ltd (SWX: ATLN) announced today that Tracleer® (bosentan), a dual endothelin receptor antagonist, has been approved in the European Union for the treatment of patients with mildly symptomatic pulmonary arterial hypertension (PAH WHO functional class FC II). Since 2002, Tracleer® has been approved and available in the European Union for PAH patients with WHO FC III.
Tracleer® is the first PAH treatment ever to be investigated in a clinical study that exclusively enrolled patients with mildly symptomatic WHO FC II. This 185-patient randomized, double-blind, placebo-controlled study provided the basis for this EU approval. The indication extension reads: "Some improvements have also been shown in patients with PAH WHO functional class II (see section 5.1)" 1
Jean-Paul Clozel, M.D. and Chief Executive Officer of Actelion, commented: "The EARLY study has demonstrated that even patients with mild symptoms are at risk of rapid deterioration. I am very proud that Actelion - together with the scientific community - has been able to demonstrate the important role of Tracleer® in delaying disease progression in these patients. Our dual endothelin receptor antagonist Tracleer® is the only PAH medicine to have demonstrated a delay in disease progression in three independent placebo-controlled, randomized clinical studies. Actelion will now communicate these important clinical findings to encourage early diagnosis and intervention."
The results from EARLY (Endothelin Antagonist tRial in miLdlY symptomatic PAH patients) published in "The Lancet" in June2, document the relentlessly progressive nature of PAH, even in its early stages, and highlight the need for earlier treatment and intervention in PAH management. The PAH progression was evident from the deterioration in all evaluated parameters in the placebo group, including the rate of clinical worsening events. The primary endpoints for the EARLY trial were changes in pulmonary vascular resistance (PVR) and exercise capacity as measured by a 6-minute walk test (6MWD). PAH progression was assessed by evaluating two secondary endpoints which were time to clinical worsening and change in WHO functional class.
The key results of the EARLY study were:
PVR improved significantly, with a reduction of 22.6 percent (p <0.0001) after six months of bosentan compared with placebo.
6MWD increased by a mean of 19 meters (p = 0.0758). Statistical significance was not seen in the 6MWD. However, this may reflect the fact that, on average, enrolled patients had a relatively well-preserved mean exercise capacity at baseline, which can be difficult to further improve.
A significant 77 percent risk reduction in time to clinical worsening (p = 0.011) was seen after six months of bosentan treatment compared with placebo. Time to clinical worsening was defined by symptomatic progression of PAH, hospitalization for PAH or death. Patients receiving bosentan had a lower incidence of worsening functional class (3.4 percent compared to 13.2 percent when receiving placebo,
p = 0.0285), providing further evidence of delayed PAH progression.
A subgroup of patients who received concomitant sildenafil showed improvements in PVR and 6MWD consistent with the overall results.
The safety and tolerability profile of bosentan in the EARLY study was consistent with that observed in previous placebo-controlled clinical trials.3,4
There are four WHO functional classes (FC) for PAH with class I being the least severe and class IV being the most advanced. These reflect the impact on a patient's life in terms of symptoms and physical activity. WHO FC II patients are defined as patients with PAH resulting in slight limitation of physical activity, they are comfortable at rest and ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope.5 Despite being mildly symptomatic, these patients still suffer from a severe and rapidly progressive disease.
Regulatory proceedings to extend the label for bosentan to include PAH patients in WHO FC II are ongoing in the US and other territories worldwide.
###
About Pulmonary Arterial Hypertension (PAH)
PAH is a syndrome characterized by a progressive increase in pulmonary vascular resistance (PVR) leading to right ventricular failure and premature death.6 If untreated, PAH carries a very poor prognosis with a median survival of 2.8 years after diagnosis.7
There are four WHO functional classes for PAH with class I being the least severe and class IV being the most advanced. These reflect the impact on a patient's life in terms of symptoms and physical activity. Class II patients are defined as patients with pulmonary hypertension resulting in mild limitation of physical activity, they are comfortable at rest and ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope.5
The pathogenesis of PAH involves the increased production of vasoactive compounds, such as endothelin. Endothelin is produced by the endothelial cells and is essential for maintenance of normal vascular tone and function. Tracleer® was the first in a new class of treatments for PAH known as endothelin receptor antagonists. Tracleer® is a dual antagonist as it blocks both ETA and ETB receptors preventing the deleterious effects of endothelin.
Online information on PAH is available at www.pah-info.com. PAH-info.com is part of an international PAH awareness campaign supported by Actelion Pharmaceuticals and has been created to provide information to healthcare professionals and patients.
About Tracleer® in Pulmonary Arterial Hypertension (PAH)
Tracleer® is an oral dual endothelin receptor antagonist, which is currently licensed for the treatment of PAH; in the United States in PAH Functional Class III and IV to improve exercise capacity and decrease the rate of clinical worsening and in Europe in PAH Functional Class III to improve exercise capacity and symptoms as well as PAH Functional Class II where some improvements have also been shown. In the EU, Tracleer® is also indicated to reduce the number of new digital ulcers in patients with systemic sclerosis and ongoing digital ulcer disease. Regulatory review for the inclusion of functional class II in the Tracleer® label is ongoing on a worldwide basis.
Tracleer® has been made commercially available by Actelion subsidiaries in the United States, the European Union, Japan, Australia, Canada, Switzerland and other markets worldwide since 2001. In these seven years of clinical experience, more than 50,000 patients have been treated with Tracleer®.
Other clinical studies with Tracleer® in specific patient populations
Actelion also conducted clinical trials to further describe the role of bosentan in treating PAH in specific patient populations, such as patients with congenital heart disease (with Eisenmenger syndrome; BREATHE-5) and patients infected with HIV (BREATHE-4). Study results are reflected in the Tracleer® product label. Clinical studies with bosentan in children suffering from PAH (BREATHE-3, FUTURE-1 and -2) have been conducted. A dedicated paediatric formulation with bosentan has been submitted to European Health Authority and is currently under assessment.
About Tracleer® in Digital Ulcers (DU)
DUs are a manifestation of the underlying vasculopathy which is central to the pathophysiology of systemic sclerosis (SSc) and pivotal in the development of PAH in SSc, one of the leading causes of death in SSc. Endothelin, a pathogenic mediator, is implicated in the underlying vasculopathy in SSc.
DUs can be a frequent, persistent and debilitating complication of SSc. They are caused by a reduction in the lumen of small bloody vessels that decreases blood flow to the fingers and toes causing open sores. DUs are painful, with a debilitating impact on patients' daily life, often making it impossible to work and undertake even simple day-to-day activities, particularly those associated with fingertip function. Reducing the occurrence of new DUs is an important and achievable treatment goal in SSc.
In the EU, Tracleer® is indicated to reduce the number of new digital ulcers in patients with systemic sclerosis and ongoing digital ulcer disease. Tracleer® has been shown to improve hand function (i.e. dressing and hygiene) in patients with scleroderma-induced digital ulcers.
Requires attention to two significant safety concerns: Potential for serious liver injury (including rare cases of liver failure and unexplained hepatic cirrhosis in a setting of close monitoring) - Liver monitoring of all patients is essential prior to initiation of treatment and monthly thereafter. High potential for major birth defects - pregnancy must be excluded and prevented by two forms of birth control; monthly pregnancy tests should be obtained. Because of these risks, Tracleer® is only supplied through a controlled distribution.
References
1. Tracleer® SPC
2. Galiè N, Rubin LJ, Hoeper MM et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008;371: 2093-2100.
3. Channick RN, Simonneau G, Sitbon O et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001;358:1119-23 (Study 351).
4. Rubin LJ, Badesch DB, Barst RJ et al. Bosentan therapy for pulmonary arterial hypertension. NEJM 2002;346:896-903 (BREATHE-1).
5. Barst RJ, McGoon M, Torbicki A et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43 (Suppl S):40S-47S.
6. Sitbon O, Humbert M, Jais X et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111:3105-3111.
7. D'Alonzo GE, Barst RJ, Ayres SM et al. Survival in patients with primary pulmonary hypertension: Results from a national prospective registry. Ann Intern Med 1991;115:343-349.
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion's first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer® through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium - the single layer of cells separating every blood vessel from the blood stream. Actelion's over 1700 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SWX Swiss Exchange (ticker symbol: ATLN).
For further information please contact:
Medical Media Contact
Kris Matta-Noaman
Packer Forbes
+44 208 772 1551
kris@packerforbes.com
Investor Contact
Roland Haefeli
Vice President, Head of Investor Relations & Public Affairs
Actelion Pharmaceuticals Ltd, Gewerbestrasse 16, CH-4123 Allschwil
+41 61 565 62 62
+1 650 624 69 36
http://www.actelion.com

Published: 07:00 04.08.2008 GMT+2 /HUGIN /Source: Actelion Pharmaceuticals Ltd /SWX: ATLN /ISIN: CH0010532478
Tracleer® (bosentan) receives EU approval for treatment of patients with mildly symptomatic Pulmonary Arterial Hypertension
ALLSCHWIL/BASEL, SWITZERLAND - 04 August 2008 - Actelion Ltd (SWX: ATLN) announced today that Tracleer® (bosentan), a dual endothelin receptor antagonist, has been approved in the European Union for the treatment of patients with mildly symptomatic pulmonary arterial hypertension (PAH WHO functional class FC II). Since 2002, Tracleer® has been approved and available in the European Union for PAH patients with WHO FC III.
Tracleer® is the first PAH treatment ever to be investigated in a clinical study that exclusively enrolled patients with mildly symptomatic WHO FC II. This 185-patient randomized, double-blind, placebo-controlled study provided the basis for this EU approval. The indication extension reads: "Some improvements have also been shown in patients with PAH WHO functional class II (see section 5.1)" 1
Jean-Paul Clozel, M.D. and Chief Executive Officer of Actelion, commented: "The EARLY study has demonstrated that even patients with mild symptoms are at risk of rapid deterioration. I am very proud that Actelion - together with the scientific community - has been able to demonstrate the important role of Tracleer® in delaying disease progression in these patients. Our dual endothelin receptor antagonist Tracleer® is the only PAH medicine to have demonstrated a delay in disease progression in three independent placebo-controlled, randomized clinical studies. Actelion will now communicate these important clinical findings to encourage early diagnosis and intervention."
The results from EARLY (Endothelin Antagonist tRial in miLdlY symptomatic PAH patients) published in "The Lancet" in June2, document the relentlessly progressive nature of PAH, even in its early stages, and highlight the need for earlier treatment and intervention in PAH management. The PAH progression was evident from the deterioration in all evaluated parameters in the placebo group, including the rate of clinical worsening events. The primary endpoints for the EARLY trial were changes in pulmonary vascular resistance (PVR) and exercise capacity as measured by a 6-minute walk test (6MWD). PAH progression was assessed by evaluating two secondary endpoints which were time to clinical worsening and change in WHO functional class.
The key results of the EARLY study were:
PVR improved significantly, with a reduction of 22.6 percent (p <0.0001) after six months of bosentan compared with placebo.
6MWD increased by a mean of 19 meters (p = 0.0758). Statistical significance was not seen in the 6MWD. However, this may reflect the fact that, on average, enrolled patients had a relatively well-preserved mean exercise capacity at baseline, which can be difficult to further improve.
A significant 77 percent risk reduction in time to clinical worsening (p = 0.011) was seen after six months of bosentan treatment compared with placebo. Time to clinical worsening was defined by symptomatic progression of PAH, hospitalization for PAH or death. Patients receiving bosentan had a lower incidence of worsening functional class (3.4 percent compared to 13.2 percent when receiving placebo,
p = 0.0285), providing further evidence of delayed PAH progression.
A subgroup of patients who received concomitant sildenafil showed improvements in PVR and 6MWD consistent with the overall results.
The safety and tolerability profile of bosentan in the EARLY study was consistent with that observed in previous placebo-controlled clinical trials.3,4
There are four WHO functional classes (FC) for PAH with class I being the least severe and class IV being the most advanced. These reflect the impact on a patient's life in terms of symptoms and physical activity. WHO FC II patients are defined as patients with PAH resulting in slight limitation of physical activity, they are comfortable at rest and ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope.5 Despite being mildly symptomatic, these patients still suffer from a severe and rapidly progressive disease.
Regulatory proceedings to extend the label for bosentan to include PAH patients in WHO FC II are ongoing in the US and other territories worldwide.
###
About Pulmonary Arterial Hypertension (PAH)
PAH is a syndrome characterized by a progressive increase in pulmonary vascular resistance (PVR) leading to right ventricular failure and premature death.6 If untreated, PAH carries a very poor prognosis with a median survival of 2.8 years after diagnosis.7
There are four WHO functional classes for PAH with class I being the least severe and class IV being the most advanced. These reflect the impact on a patient's life in terms of symptoms and physical activity. Class II patients are defined as patients with pulmonary hypertension resulting in mild limitation of physical activity, they are comfortable at rest and ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope.5
The pathogenesis of PAH involves the increased production of vasoactive compounds, such as endothelin. Endothelin is produced by the endothelial cells and is essential for maintenance of normal vascular tone and function. Tracleer® was the first in a new class of treatments for PAH known as endothelin receptor antagonists. Tracleer® is a dual antagonist as it blocks both ETA and ETB receptors preventing the deleterious effects of endothelin.
Online information on PAH is available at www.pah-info.com. PAH-info.com is part of an international PAH awareness campaign supported by Actelion Pharmaceuticals and has been created to provide information to healthcare professionals and patients.
About Tracleer® in Pulmonary Arterial Hypertension (PAH)
Tracleer® is an oral dual endothelin receptor antagonist, which is currently licensed for the treatment of PAH; in the United States in PAH Functional Class III and IV to improve exercise capacity and decrease the rate of clinical worsening and in Europe in PAH Functional Class III to improve exercise capacity and symptoms as well as PAH Functional Class II where some improvements have also been shown. In the EU, Tracleer® is also indicated to reduce the number of new digital ulcers in patients with systemic sclerosis and ongoing digital ulcer disease. Regulatory review for the inclusion of functional class II in the Tracleer® label is ongoing on a worldwide basis.
Tracleer® has been made commercially available by Actelion subsidiaries in the United States, the European Union, Japan, Australia, Canada, Switzerland and other markets worldwide since 2001. In these seven years of clinical experience, more than 50,000 patients have been treated with Tracleer®.
Other clinical studies with Tracleer® in specific patient populations
Actelion also conducted clinical trials to further describe the role of bosentan in treating PAH in specific patient populations, such as patients with congenital heart disease (with Eisenmenger syndrome; BREATHE-5) and patients infected with HIV (BREATHE-4). Study results are reflected in the Tracleer® product label. Clinical studies with bosentan in children suffering from PAH (BREATHE-3, FUTURE-1 and -2) have been conducted. A dedicated paediatric formulation with bosentan has been submitted to European Health Authority and is currently under assessment.
About Tracleer® in Digital Ulcers (DU)
DUs are a manifestation of the underlying vasculopathy which is central to the pathophysiology of systemic sclerosis (SSc) and pivotal in the development of PAH in SSc, one of the leading causes of death in SSc. Endothelin, a pathogenic mediator, is implicated in the underlying vasculopathy in SSc.
DUs can be a frequent, persistent and debilitating complication of SSc. They are caused by a reduction in the lumen of small bloody vessels that decreases blood flow to the fingers and toes causing open sores. DUs are painful, with a debilitating impact on patients' daily life, often making it impossible to work and undertake even simple day-to-day activities, particularly those associated with fingertip function. Reducing the occurrence of new DUs is an important and achievable treatment goal in SSc.
In the EU, Tracleer® is indicated to reduce the number of new digital ulcers in patients with systemic sclerosis and ongoing digital ulcer disease. Tracleer® has been shown to improve hand function (i.e. dressing and hygiene) in patients with scleroderma-induced digital ulcers.
Requires attention to two significant safety concerns: Potential for serious liver injury (including rare cases of liver failure and unexplained hepatic cirrhosis in a setting of close monitoring) - Liver monitoring of all patients is essential prior to initiation of treatment and monthly thereafter. High potential for major birth defects - pregnancy must be excluded and prevented by two forms of birth control; monthly pregnancy tests should be obtained. Because of these risks, Tracleer® is only supplied through a controlled distribution.
References
1. Tracleer® SPC
2. Galiè N, Rubin LJ, Hoeper MM et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008;371: 2093-2100.
3. Channick RN, Simonneau G, Sitbon O et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001;358:1119-23 (Study 351).
4. Rubin LJ, Badesch DB, Barst RJ et al. Bosentan therapy for pulmonary arterial hypertension. NEJM 2002;346:896-903 (BREATHE-1).
5. Barst RJ, McGoon M, Torbicki A et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43 (Suppl S):40S-47S.
6. Sitbon O, Humbert M, Jais X et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111:3105-3111.
7. D'Alonzo GE, Barst RJ, Ayres SM et al. Survival in patients with primary pulmonary hypertension: Results from a national prospective registry. Ann Intern Med 1991;115:343-349.
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion's first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer® through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium - the single layer of cells separating every blood vessel from the blood stream. Actelion's over 1700 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SWX Swiss Exchange (ticker symbol: ATLN).
For further information please contact:
Medical Media Contact
Kris Matta-Noaman
Packer Forbes
+44 208 772 1551
kris@packerforbes.com
Investor Contact
Roland Haefeli
Vice President, Head of Investor Relations & Public Affairs
Actelion Pharmaceuticals Ltd, Gewerbestrasse 16, CH-4123 Allschwil
+41 61 565 62 62
+1 650 624 69 36
http://www.actelion.com
Antwort auf Beitrag Nr.: 34.643.067 von Fruehrentner am 04.08.08 15:08:52off topic... nur eben, da ich Actelion nicht im Depot habe:
Zürich (aktiencheck.de AG) - Andrew C. Weiss, Analyst von Vontobel Research, bewertet die Aktie von Actelion (ISIN CH0010532478 / WKN 936767) in der aktuellen Ausgabe von "Vontobel Morning Focus" nach wie vor mit "hold".
Actelion habe heute Morgen bekannt gegeben, aufgrund der EARLY-Daten die Zulassung für Tracleer der Funktionsklasse II erhalten zu haben. Tracleer habe bereits die Zulassung für die Behandlung der Funktionsklasse III und IV-PAH-Patienten (Patienten mit leichter symptomatischer pulmonaler arterieller Hypertonie) erhalten. Das Pricing sei also bereits verhandelt worden.
Die EU-Zulassung sei erwartet worden, da das CHMP Ende Juni eine positive Stellungnahme abgegeben habe, aber die Zulassung erfolge etwas schneller als die Analysten gedacht hätten. Sie seien von einer EU-Zulassung im September 2008 ausgegangen.
Die Daten der EARLY-Studie seien im Juni 2008 in der Fachzeitschrift "The Lancet" veröffentlicht worden. Diese Studiendaten hätten dazu gedient, die Indikationserweiterung für Tracleer zu erhalten, und es sei die einzige Studie, die den Einsatz eines Endothelin-Rezeptor-Antagonisten (in diesem Fall Tracleer) ausschließlich bei PAH der Funktionsklasse II untersucht habe.
Mit dieser Zulassung erhalte Actelion Zugang zu einem neuen Pool möglicher Patienten, die eine PAH-Behandlung benötigen würden. Der Abnehmermarkt könnte sich möglicherweise verdoppeln. Aber da die Patienten nur leichte Symptome zeigen würden, werde es schwierig sein, diese neuen Patienten zu finden, eine solide ärztliche Ausbildung werde erforderlich sein. Die Analysten würden derzeit auf aktuelle Informationen von der US-Gesundheitsbehörde FDA bezüglich der EARLY-Daten zum Tracleer-Label warten.
Die Zulassung der EARLY-Daten für das Tracleer-Label sei weitherum erwartet worden. Die Analysten würden bezüglich Marktakzeptanz von Tracleer konservative Schätzungen machen, was einer langsamen Marktdurchdringung bei dieser Indikation entspreche, zumal das Finden von Patienten eine hohe ärztliche Ausbildung erfordere. Zuvor hätten sie auch von Gilead erwartet, dass der Konzern bei der Vermarktung von Letairis größere Anstrengungen unternehmen und so zur Markterweiterung von Actelion beitragen würde. Doch angesichts der schwachen Marketing-Unterstützung würden sie davon ausgehen, dass Actelion die meiste Knochenarbeit bei der ärztlichen Ausbildung selbst erledigen müsse.
mfg ipollit
Zürich (aktiencheck.de AG) - Andrew C. Weiss, Analyst von Vontobel Research, bewertet die Aktie von Actelion (ISIN CH0010532478 / WKN 936767) in der aktuellen Ausgabe von "Vontobel Morning Focus" nach wie vor mit "hold".
Actelion habe heute Morgen bekannt gegeben, aufgrund der EARLY-Daten die Zulassung für Tracleer der Funktionsklasse II erhalten zu haben. Tracleer habe bereits die Zulassung für die Behandlung der Funktionsklasse III und IV-PAH-Patienten (Patienten mit leichter symptomatischer pulmonaler arterieller Hypertonie) erhalten. Das Pricing sei also bereits verhandelt worden.
Die EU-Zulassung sei erwartet worden, da das CHMP Ende Juni eine positive Stellungnahme abgegeben habe, aber die Zulassung erfolge etwas schneller als die Analysten gedacht hätten. Sie seien von einer EU-Zulassung im September 2008 ausgegangen.
Die Daten der EARLY-Studie seien im Juni 2008 in der Fachzeitschrift "The Lancet" veröffentlicht worden. Diese Studiendaten hätten dazu gedient, die Indikationserweiterung für Tracleer zu erhalten, und es sei die einzige Studie, die den Einsatz eines Endothelin-Rezeptor-Antagonisten (in diesem Fall Tracleer) ausschließlich bei PAH der Funktionsklasse II untersucht habe.
Mit dieser Zulassung erhalte Actelion Zugang zu einem neuen Pool möglicher Patienten, die eine PAH-Behandlung benötigen würden. Der Abnehmermarkt könnte sich möglicherweise verdoppeln. Aber da die Patienten nur leichte Symptome zeigen würden, werde es schwierig sein, diese neuen Patienten zu finden, eine solide ärztliche Ausbildung werde erforderlich sein. Die Analysten würden derzeit auf aktuelle Informationen von der US-Gesundheitsbehörde FDA bezüglich der EARLY-Daten zum Tracleer-Label warten.
Die Zulassung der EARLY-Daten für das Tracleer-Label sei weitherum erwartet worden. Die Analysten würden bezüglich Marktakzeptanz von Tracleer konservative Schätzungen machen, was einer langsamen Marktdurchdringung bei dieser Indikation entspreche, zumal das Finden von Patienten eine hohe ärztliche Ausbildung erfordere. Zuvor hätten sie auch von Gilead erwartet, dass der Konzern bei der Vermarktung von Letairis größere Anstrengungen unternehmen und so zur Markterweiterung von Actelion beitragen würde. Doch angesichts der schwachen Marketing-Unterstützung würden sie davon ausgehen, dass Actelion die meiste Knochenarbeit bei der ärztlichen Ausbildung selbst erledigen müsse.
mfg ipollit
Regeneron - REGN
http://www.regeneron.com/
Marktkapitalisierung: 1,73 Mrd USD
Cash: 743 Mio USD
Schulden: 200 Mio USD
Umsatz 2008e/2009e: 222 / 240 Mio USD
Gewinn: (keine Gewinne)
zugelassene Produkte: ARCALYST
Partner: u.a. Sanofi Aventis (ist zu 19% an Regeneron beteiligt), Bayer
Technologien: AK, Trap
Regeneron ist eines der ältesten Biotechs ähnlich wie Amgen oder Genentech, war aber bei weitem nicht so erfolgreich.
Pipeline:
Aflibercept (VEGF-Trap):
Es handelt sich dabei um einen sogenannten Zytokin-Fänger (Trap), wenn ich das richtig übersetzt habe. Damit werden ähnlich wie mit z.B. Antikörpern Signalmoleküle eingefangen und somit davon abgehalten, ihren spezifischen Rezeptor zu aktivieren, der irgendetwas auslöst. Im Falle von VEGF-Trap wird der Wachstumsfaktor VEGF-A gebunden, den Krebszellen benötigen, um ihre Blutversorgung wachsen zu lassen. Damit basiert Aflibercept auf dem gleichen Wirkprinzip wie der Antikörper Avastin und steht damit in dessen Konkurrenz. Aflibercept ist mit Sanofi verpartnert, die die Entwicklung mit finanzieren. Indikationen sind alle Krebsarten, bei denen Avastin ebenfalls zu Einsatz kommt (Avastin ist das erfolgreichste Krebsmedikament mit mehreren Mrd USD Umsatz). Die Pipeline unten ist nicht ganz vollständig. Aktuelle Studien sind:
PIII-Studien:
- first-line metastatic hormone resistant prostate cancer in combination with taxotere/prednisone
- second-line non-small cell lung cancer in combination with taxotere
- first-line metastatic pancreatic cancer in combination with gemcitabine-based regimen
- second-line metastatic colorectal cancer in combination with FOLFIRI (Folinic Acid, Fluorouracil and irinotecan)
Monotherapie PIIs:
- Advanced ovarian cancer (AOC)
- Non-small cell lung adenocarcinoma (NSCLA)
- Advanced ovarian cancer patients with symptomatic malignant ascites (SMA)
weitere PIIs:
- Metastatic breast cancer
- Metastatic or unresectable kidney cancer
- Recurrent ovarian cancer
- Recurrent malignant gliomas
- Relapsed/refractory multiple myeloma
- Metastatic colorectal cancer
VEGF Trap Eye:
Ebenfalls ein Trap, der VEGF-A und PLGF bindet. Ähnlich wie Genentech's Lucentis soll es bei AMD (altersbedingte Veränderungen der Netzhaut im Auge, die zur Blindheit führen), sowie Varianten davon zum Einsatz kommen. Der Partner ist in diesem Fall Bayer. Gegen wet AMD befindet es sich in PIII, DME (im Zusammenhang mit Diabetes) ist ebenfalls geplant.
Rilonacept:
Ist gegen eine seltene Erkrankung als ARCALYST bereits zugelassen. Wird zurzeit gegen die größere Indikation der Gicht in PII getestet.
REGN88:
Ist der erste Antikörper-Kandidat, der von der umfangreichen Kooperation mit Sanofi in die Klinik gegangen ist. Er richtet sich gegen IL-6 und soll bei Rheumatischer Arthritis angewendet werden. Entwickelt wurde er mit der REGN-Technologie VelocImmune, die spezielle Knockout-Mäuse für die Antikörper-Gewinnung verwendet.

29.11.2007 11:22
Sanofi-Aventis will Regeneron-Beteiligung auf 19% ausweiten
DJ Sanofi-Aventis (News/Aktienkurs) will Regeneron-Beteiligung auf 19% ausweiten
PARIS (Dow Jones)--Der Pharmakonzern Sanofi-Aventis SA will seinen Anteil am US-Konkurrenten Regeneron Pharmaceuticals Inc (News) auf 19% von aktuell 4% erhöhen. Zu diesem Zweck sollen 12 Mio neu emittierte Regeneron-Aktien zum Stückpreis von 26 USD gekauft werden, teilte die in Paris ansässige Sanofi-Aventis am Donnerstag mit. Die Transaktion stehe noch unter dem Vorbehalt der Zustimmung der Kartellbehörden.
Beide Unternehmen haben zudem eine Kooperationsvereinbarung zur gemeinsamen Entwicklung therapeutischer Antikörper unterzeichnet. Sanofi wird Regeneron demnach in den kommenden fünf Jahren insgesamt 85 Mio USD zahlen. Hinzu komme eine Zahlung von bis zu 475 Mio USD, die in den Forschungsetat des US-Konzerns fließen wird.
Im Gegenzug werde der in den USA erzielte Gewinn hälftig unter beiden Pharmakonzernen aufgeteilt. Der in anderen Ländern anfallende Gewinn werde künftig entsprechend vertraglich geregelten Konditionen verteilt.
mfg ipollit
http://www.regeneron.com/
Marktkapitalisierung: 1,73 Mrd USD
Cash: 743 Mio USD
Schulden: 200 Mio USD
Umsatz 2008e/2009e: 222 / 240 Mio USD
Gewinn: (keine Gewinne)
zugelassene Produkte: ARCALYST
Partner: u.a. Sanofi Aventis (ist zu 19% an Regeneron beteiligt), Bayer
Technologien: AK, Trap
Regeneron ist eines der ältesten Biotechs ähnlich wie Amgen oder Genentech, war aber bei weitem nicht so erfolgreich.
Pipeline:
Aflibercept (VEGF-Trap):
Es handelt sich dabei um einen sogenannten Zytokin-Fänger (Trap), wenn ich das richtig übersetzt habe. Damit werden ähnlich wie mit z.B. Antikörpern Signalmoleküle eingefangen und somit davon abgehalten, ihren spezifischen Rezeptor zu aktivieren, der irgendetwas auslöst. Im Falle von VEGF-Trap wird der Wachstumsfaktor VEGF-A gebunden, den Krebszellen benötigen, um ihre Blutversorgung wachsen zu lassen. Damit basiert Aflibercept auf dem gleichen Wirkprinzip wie der Antikörper Avastin und steht damit in dessen Konkurrenz. Aflibercept ist mit Sanofi verpartnert, die die Entwicklung mit finanzieren. Indikationen sind alle Krebsarten, bei denen Avastin ebenfalls zu Einsatz kommt (Avastin ist das erfolgreichste Krebsmedikament mit mehreren Mrd USD Umsatz). Die Pipeline unten ist nicht ganz vollständig. Aktuelle Studien sind:
PIII-Studien:
- first-line metastatic hormone resistant prostate cancer in combination with taxotere/prednisone
- second-line non-small cell lung cancer in combination with taxotere
- first-line metastatic pancreatic cancer in combination with gemcitabine-based regimen
- second-line metastatic colorectal cancer in combination with FOLFIRI (Folinic Acid, Fluorouracil and irinotecan)
Monotherapie PIIs:
- Advanced ovarian cancer (AOC)
- Non-small cell lung adenocarcinoma (NSCLA)
- Advanced ovarian cancer patients with symptomatic malignant ascites (SMA)
weitere PIIs:
- Metastatic breast cancer
- Metastatic or unresectable kidney cancer
- Recurrent ovarian cancer
- Recurrent malignant gliomas
- Relapsed/refractory multiple myeloma
- Metastatic colorectal cancer
VEGF Trap Eye:
Ebenfalls ein Trap, der VEGF-A und PLGF bindet. Ähnlich wie Genentech's Lucentis soll es bei AMD (altersbedingte Veränderungen der Netzhaut im Auge, die zur Blindheit führen), sowie Varianten davon zum Einsatz kommen. Der Partner ist in diesem Fall Bayer. Gegen wet AMD befindet es sich in PIII, DME (im Zusammenhang mit Diabetes) ist ebenfalls geplant.
Rilonacept:
Ist gegen eine seltene Erkrankung als ARCALYST bereits zugelassen. Wird zurzeit gegen die größere Indikation der Gicht in PII getestet.
REGN88:
Ist der erste Antikörper-Kandidat, der von der umfangreichen Kooperation mit Sanofi in die Klinik gegangen ist. Er richtet sich gegen IL-6 und soll bei Rheumatischer Arthritis angewendet werden. Entwickelt wurde er mit der REGN-Technologie VelocImmune, die spezielle Knockout-Mäuse für die Antikörper-Gewinnung verwendet.
29.11.2007 11:22
Sanofi-Aventis will Regeneron-Beteiligung auf 19% ausweiten
DJ Sanofi-Aventis (News/Aktienkurs) will Regeneron-Beteiligung auf 19% ausweiten
PARIS (Dow Jones)--Der Pharmakonzern Sanofi-Aventis SA will seinen Anteil am US-Konkurrenten Regeneron Pharmaceuticals Inc (News) auf 19% von aktuell 4% erhöhen. Zu diesem Zweck sollen 12 Mio neu emittierte Regeneron-Aktien zum Stückpreis von 26 USD gekauft werden, teilte die in Paris ansässige Sanofi-Aventis am Donnerstag mit. Die Transaktion stehe noch unter dem Vorbehalt der Zustimmung der Kartellbehörden.
Beide Unternehmen haben zudem eine Kooperationsvereinbarung zur gemeinsamen Entwicklung therapeutischer Antikörper unterzeichnet. Sanofi wird Regeneron demnach in den kommenden fünf Jahren insgesamt 85 Mio USD zahlen. Hinzu komme eine Zahlung von bis zu 475 Mio USD, die in den Forschungsetat des US-Konzerns fließen wird.
Im Gegenzug werde der in den USA erzielte Gewinn hälftig unter beiden Pharmakonzernen aufgeteilt. Der in anderen Ländern anfallende Gewinn werde künftig entsprechend vertraglich geregelten Konditionen verteilt.
mfg ipollit
Änderungen im Depot...
- weitere Reduzierung der Regeneron-Position; REGN ist mir doch zu riskant, um so deutlich übergewichtet zu sein. Ziel sind etwa 10% Depotanteil.
- weitere Reduzierung der Imclone-Position; IMCL steht mit ca. 65 USD deutlich über den gebotenen 60 USD. Ich kann nicht abschätzen, wieviel ein verbessertes Angebot erzielen kann... vielleicht nochmal nochmal 10% über dem aktuellen Kurs? Aber garantiert ist es nicht und der Zeitpunkt ist ebenfalls unklar.
- Aufstockung der Evotec-Position
- Aufbau von Positionen in Rigel (aussichtsreiches orales RA-Mittel in PII) und Incyte (JAK-Hemmer in PII): beide Unternehmen bereits hoch bewertet, zwar mit Chancen aber auch mit dem Risiko eines deutlichen Rückschlags. Daher nur spekulative kleine Positionen.
23,5% Genmab http://finance.yahoo.com/q?s=GEN.CO
13,5% Regeneron http://finance.yahoo.com/q?s=REGN
9,1% Onyx http://finance.yahoo.com/q?s=ONXX
7,0% Evotec http://finance.yahoo.com/q?s=EVT.DE
6,8% OSI Pharma http://finance.yahoo.com/q?s=OSIP
5,9% (KO) Intercell http://finance.yahoo.com/q?s=IJE.F
5,4% Cubist http://finance.yahoo.com/q?s=CBST
5,0% Isis http://finance.yahoo.com/q?s=ISIS
4,8% Vertex http://finance.yahoo.com/q?s=VRTX
3,4% Arena http://finance.yahoo.com/q?s=ARNA
3,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,0% Micromet http://finance.yahoo.com/q?s=MITI
2,6% Rigel http://finance.yahoo.com/q?s=RIGL
2,6% Imclone http://finance.yahoo.com/q?s=IMCL
2,5% Incyte http://finance.yahoo.com/q?s=INCY
1,8% Supergen http://finance.yahoo.com/q?s=SUPG
Watchliste: POZN (Spekulation auf keinen völligen Fehlschlag bei der Vermarktung von Treximent, sowie dem Erfolg von PN 400), SGEN (hoch bewertet, scheint aber ein wesentlicher Teil der Genentech-Pipeline zu bilden und die AK-Technologie könnte Potential haben) und MNTA (Generika-Technologie)
mfg ipollit
- weitere Reduzierung der Regeneron-Position; REGN ist mir doch zu riskant, um so deutlich übergewichtet zu sein. Ziel sind etwa 10% Depotanteil.
- weitere Reduzierung der Imclone-Position; IMCL steht mit ca. 65 USD deutlich über den gebotenen 60 USD. Ich kann nicht abschätzen, wieviel ein verbessertes Angebot erzielen kann... vielleicht nochmal nochmal 10% über dem aktuellen Kurs? Aber garantiert ist es nicht und der Zeitpunkt ist ebenfalls unklar.
- Aufstockung der Evotec-Position
- Aufbau von Positionen in Rigel (aussichtsreiches orales RA-Mittel in PII) und Incyte (JAK-Hemmer in PII): beide Unternehmen bereits hoch bewertet, zwar mit Chancen aber auch mit dem Risiko eines deutlichen Rückschlags. Daher nur spekulative kleine Positionen.
23,5% Genmab http://finance.yahoo.com/q?s=GEN.CO
13,5% Regeneron http://finance.yahoo.com/q?s=REGN
9,1% Onyx http://finance.yahoo.com/q?s=ONXX
7,0% Evotec http://finance.yahoo.com/q?s=EVT.DE
6,8% OSI Pharma http://finance.yahoo.com/q?s=OSIP
5,9% (KO) Intercell http://finance.yahoo.com/q?s=IJE.F
5,4% Cubist http://finance.yahoo.com/q?s=CBST
5,0% Isis http://finance.yahoo.com/q?s=ISIS
4,8% Vertex http://finance.yahoo.com/q?s=VRTX
3,4% Arena http://finance.yahoo.com/q?s=ARNA
3,1% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,0% Micromet http://finance.yahoo.com/q?s=MITI
2,6% Rigel http://finance.yahoo.com/q?s=RIGL
2,6% Imclone http://finance.yahoo.com/q?s=IMCL
2,5% Incyte http://finance.yahoo.com/q?s=INCY
1,8% Supergen http://finance.yahoo.com/q?s=SUPG
Watchliste: POZN (Spekulation auf keinen völligen Fehlschlag bei der Vermarktung von Treximent, sowie dem Erfolg von PN 400), SGEN (hoch bewertet, scheint aber ein wesentlicher Teil der Genentech-Pipeline zu bilden und die AK-Technologie könnte Potential haben) und MNTA (Generika-Technologie)
mfg ipollit
zu Genmab...
die aktuell starke Übergewichtung halte ich für gerechtfertigt, da sich in der Pipeline u.a. mit HuMax-CD20 eine vielversprechende Alternative zum etablierten Blockbuster Rituxan und mit HuMax-Egfr eine Alternative zum Blockbuster Erbitux in der fortgeschrittenen PIII befindet. Rituxan ist eine wesentliche Basis von BiogenIdec (MK 14 Mrd USD) und Erbitux ist Imclone (MK 5,5 Mrd USD). Zudem besteht Übernahme-Phantasie durch GSK.
Im Analystenkommentar unten:
- in der PIII von HuMax-CD20 deutlich bessere Daten (51% und 44% RR) beim refraktionärem CLL als von Rituxan (25% RR) und Campath (33% RR)
- mit fasttrack Zulassung bereits Anfang 2009 möglich; dies würde einen 200 Mio USD Meilenstein an Genmab auslösen
- off-label Einsatz geplant
- GSK soll Produktionskapazitäten reserviert haben, um HuMax-CD20 im Wert von 1,2 bis 2 Mrd USD produzieren zu können
- HuMax-Egfr PIII-Daten bald verfügbar, die für eine Zulassung ausreichen könnten (HuMax-EGFR ist noch nicht verpartnert! Volle Rechte bei Genmab)
********
Bullets Genmab: Spectacular Phase III data HuMax CD20 CLL, BUY maintained by SNS Securities
Yesterday, Genmab announced spectacular Phase III data of HuMax CD20 (ofatumumab) CLL in refractory patients. The overall response rate of the two patients group treated was 51% in patients refractory to fludarabine (chemotherapy) and Campath (competitive antibody against CLL), and 44% in patients to fludarabine alone. Market consensus was around 25% overall response rate.
Also compared to Phase III data of competitive antibodies like Rituxan (25% response rate in refractory CLL patients) and Campath (33% response rate), the data of Genmab are superior. In our view there should be no doubt that this product will be approved by the FDA. We expect Genmab/GSK to go for BLA filing very soon. The filing and the subsequent approval will trigger a milestone that we estimate to be USD 200m. Genmab has a fast track on HuMax CD20 CLL, so the FDA is likely to give approval between 2 and 6 months.
The strong Phase III data on refractory patients also makes a strong case for the off label use of the drug for other groups of patients (first line) and even for other indications like Non Hodgkin’s Lymphoma. It has always been Genmab’s strategy to go for off label use after getting the Phase III data on HuMax CD20 CLL in refractory patients. In the next few weeks, Genmab/GSK will likely start a large Phase III study with HuMax CD20 in first line patients. The first data on this study should coincide with the market launch of the drug in refractory patients. Positive data would support the launch of the product, which we expect to happen end 2009Q1.
The case of a strong and swift market introduction is backed by GSK, who seems to have made reservations on 40,000 liter capacity at Lonza for the production of HuMax CD20. Considering a yield of 3-5 grams per liter, this would mean USD 1.2-2bn.
Later this year, an interim analysis of HuMax EGFr refractory H&N Phase III is to be expected. In April 132 patients were included in the pivotal trial. The last few weeks patient inclusion is about 5-10 persons per week. Counted from April this would mean that now 192-252 patients are enrolled. Two third of patients are receiving the drug. An interim analysis is possible with the data of 116 patients. The longer it takes that we need to wait for the interim analysis, the better the data are expected to be (with higher survival rates, current treatment in refractory has survival rate of about 3-4 months). The recent bid on ImClone by BMS (USD 4.5bn, specifically on its drug Erbitux), also gives an indication what the potential value is for this program.
We believe that the strong data on HuMax CD20 CLL will pave the way for a move from GSK in taking over the company. The time frame seems to be between August and November (between CLL data and interim analysis EGFr). With positive data on CLL, GSK has the hard data in hand to validate a take over. Wating till the announcement of the interim analysis on HuMax EGFr H&N would only increase the price. Pressure to come with interim analysis on EGFr sooner rather than later will increase in the next few months. Good data on 116 patients should be enough to file for approval. For GSK the risks have decreased considerably with the hard data on HuMax CD20. Also bear in mind that for GSK the financial ratio to go for a take over scenario becomes more interesting, since the company still needs to pay milestones up to USD 1bn and royalties up to 50%.
A take over scenario is becoming also more likely considering the ongoing M&A activity in the sector. The biotech companies that have been taken over announced positive Phase III data and /or already have products on the market. Genmab now can also be put in this select group of candidates.
In all, we strongly maintain our Buy rating on the stock. We advice our clients to maintain current holdings or even increase stakes in the company. At the current share price we still see considerable upside potential of 60-100%.
Regards,
Marcel Wijma
Senior Equity Analyst Biotechnology
SNS Securities N.V.
mfg ipollit
die aktuell starke Übergewichtung halte ich für gerechtfertigt, da sich in der Pipeline u.a. mit HuMax-CD20 eine vielversprechende Alternative zum etablierten Blockbuster Rituxan und mit HuMax-Egfr eine Alternative zum Blockbuster Erbitux in der fortgeschrittenen PIII befindet. Rituxan ist eine wesentliche Basis von BiogenIdec (MK 14 Mrd USD) und Erbitux ist Imclone (MK 5,5 Mrd USD). Zudem besteht Übernahme-Phantasie durch GSK.
Im Analystenkommentar unten:
- in der PIII von HuMax-CD20 deutlich bessere Daten (51% und 44% RR) beim refraktionärem CLL als von Rituxan (25% RR) und Campath (33% RR)
- mit fasttrack Zulassung bereits Anfang 2009 möglich; dies würde einen 200 Mio USD Meilenstein an Genmab auslösen
- off-label Einsatz geplant
- GSK soll Produktionskapazitäten reserviert haben, um HuMax-CD20 im Wert von 1,2 bis 2 Mrd USD produzieren zu können
- HuMax-Egfr PIII-Daten bald verfügbar, die für eine Zulassung ausreichen könnten (HuMax-EGFR ist noch nicht verpartnert! Volle Rechte bei Genmab)
********
Bullets Genmab: Spectacular Phase III data HuMax CD20 CLL, BUY maintained by SNS Securities
Yesterday, Genmab announced spectacular Phase III data of HuMax CD20 (ofatumumab) CLL in refractory patients. The overall response rate of the two patients group treated was 51% in patients refractory to fludarabine (chemotherapy) and Campath (competitive antibody against CLL), and 44% in patients to fludarabine alone. Market consensus was around 25% overall response rate.
Also compared to Phase III data of competitive antibodies like Rituxan (25% response rate in refractory CLL patients) and Campath (33% response rate), the data of Genmab are superior. In our view there should be no doubt that this product will be approved by the FDA. We expect Genmab/GSK to go for BLA filing very soon. The filing and the subsequent approval will trigger a milestone that we estimate to be USD 200m. Genmab has a fast track on HuMax CD20 CLL, so the FDA is likely to give approval between 2 and 6 months.
The strong Phase III data on refractory patients also makes a strong case for the off label use of the drug for other groups of patients (first line) and even for other indications like Non Hodgkin’s Lymphoma. It has always been Genmab’s strategy to go for off label use after getting the Phase III data on HuMax CD20 CLL in refractory patients. In the next few weeks, Genmab/GSK will likely start a large Phase III study with HuMax CD20 in first line patients. The first data on this study should coincide with the market launch of the drug in refractory patients. Positive data would support the launch of the product, which we expect to happen end 2009Q1.
The case of a strong and swift market introduction is backed by GSK, who seems to have made reservations on 40,000 liter capacity at Lonza for the production of HuMax CD20. Considering a yield of 3-5 grams per liter, this would mean USD 1.2-2bn.

Later this year, an interim analysis of HuMax EGFr refractory H&N Phase III is to be expected. In April 132 patients were included in the pivotal trial. The last few weeks patient inclusion is about 5-10 persons per week. Counted from April this would mean that now 192-252 patients are enrolled. Two third of patients are receiving the drug. An interim analysis is possible with the data of 116 patients. The longer it takes that we need to wait for the interim analysis, the better the data are expected to be (with higher survival rates, current treatment in refractory has survival rate of about 3-4 months). The recent bid on ImClone by BMS (USD 4.5bn, specifically on its drug Erbitux), also gives an indication what the potential value is for this program.
We believe that the strong data on HuMax CD20 CLL will pave the way for a move from GSK in taking over the company. The time frame seems to be between August and November (between CLL data and interim analysis EGFr). With positive data on CLL, GSK has the hard data in hand to validate a take over. Wating till the announcement of the interim analysis on HuMax EGFr H&N would only increase the price. Pressure to come with interim analysis on EGFr sooner rather than later will increase in the next few months. Good data on 116 patients should be enough to file for approval. For GSK the risks have decreased considerably with the hard data on HuMax CD20. Also bear in mind that for GSK the financial ratio to go for a take over scenario becomes more interesting, since the company still needs to pay milestones up to USD 1bn and royalties up to 50%.
A take over scenario is becoming also more likely considering the ongoing M&A activity in the sector. The biotech companies that have been taken over announced positive Phase III data and /or already have products on the market. Genmab now can also be put in this select group of candidates.
In all, we strongly maintain our Buy rating on the stock. We advice our clients to maintain current holdings or even increase stakes in the company. At the current share price we still see considerable upside potential of 60-100%.
Regards,
Marcel Wijma
Senior Equity Analyst Biotechnology
SNS Securities N.V.
mfg ipollit
zu Genmab's HuMax-EGFR... im Zusammenhang mit der Imclone-Übernahme durch BMS bzw. dem Wert von Erbitux.
Beim Deal scheint auch der Erbitux-Nachfolger IMC-11F8 eine wesentliche Rolle zu spielen. IMC-11F8 ist allerdings erst in PII und deutlich hinter HuMax-EGFR. Daher ist es interessant, wie IMC-11F8 hier bewertet wird:
ImClone's Value May Rest On Debate Over Next Erbitux
August 07, 2008: 10:52 AM EST
NEW YORK -(Dow Jones)- The battle for ImClone Systems Inc. (IMCL) may come down to the fight over who has rights to the follow-up to the company's sole but increasingly successful product, the cancer treatment Erbitux.
ImClone claims to own the complete rights to the next-generation Erbitux, which carries high expectations but isn't seen on the market until well into the next decade. That interpretation contradicts the company's own statement two years ago and is disputed by Bristol-Myers Squibb Co. (BMY), which co-markets the current Erbitux in the U.S. and is seeking to buy the 83% of ImClone that it doesn't own for $4.5 billion.
ImClone has said Bristol's offer "greatly undervalues" the company partly because of its product pipeline, of which the next-generation Erbitux is a key part.
Resolving the next-generation Erbitux dispute could influence billions of dollars in sales in the next decade, ImClone's total value, the action of other possible suitors and whether ImClone attempts to spin off its pipeline, a move the company said it was considering.
The disagreement centers on whether the drug, called IMC-11F8, was derived in any way from the current Erbitux and if its development can be viewed as a " competing product," as defined by the 2001 agreement between the companies. If either of those are true, Bristol is entitled to rights to the drug.
"My read is that the contract language is nebulous," Cowen & Co. analyst Eric Schmidt said. There is "clearly room for (Bristol) to claim one thing and ( ImClone) the other."
Because of the confusion, the battle over IMC-11F8 could end up in front of an arbitrator and may take years to resolve - something that Bristol-Myers can avoid by buying ImClone, which may mean raising its offer of $60 a share. Wall Street expects a higher offer for ImClone as the stock closed Wednesday at $ 64.07 and recently traded at $64.09.
The IMC-11F8 dispute comes as the first-generation Erbitux gains traction. Erbitux is approved to treat head, neck and colorectal cancer; is expected to be filed for approval in lung cancer in the fourth quarter; and is being studied in multiple other forms of the disease.
UBS projects 2008 global Erbitux sales of $1.7 billion, up 31% from $1.3 billion in 2007, and sees that rising to $3.1 billion in 2011.
Such sales estimates aren't available for IMC-11F8, which won't enter pivotal trials - likely to last at least 2 1/2 years - until the first half of 2009. The success of the first-generation Erbitux reduces some risk on the follow-up because it has a similar target. The key difference between the two is that the new drug is a human antibody, expected to be safer and have less-frequent dosing, while Erbitux is a hybrid of a human and mouse antibody.
The unknown over IMC-11F8's rights is likely to give pause to other potential suitors of ImClone or any spinoff of its pipeline. That's because other companies may be less willing to pay billions for a company if they then have to share the profits on a key future drug.
Of course, if Bristol buys ImClone, that issue would be moot, which is why some might see the recent posturing - that started from ImClone before Bristol made its offer - as a bargaining tactic.
Conflicting Views
ImClone has asserted, as recently as two weeks ago, that it owns all the rights to IMC-11F8, and in a statement this week said that Bristol-Myers "may have no rights to market" the drug.
Bristol-Myers disagrees. "We believe we have the rights to 11F8 under our existing contractual agreement," a Bristol-Myers spokeswoman said this week, with no further comments on the company's strategy for claiming the rights.
The agreement, dating from 2001, covers Erbitux, along with "all fully humanized or human" versions, analogs or derivatives of the drug.
ImClone's recent comments even conflict with its own language from April 2006, when ImClone won an arbitration decision against Erbitux's international marketing partner, Germany's Merck KGaA (MRK.XE), for the international rights related to IMC-11F8. The decision gave ImClone the rights to develop and commercialize the drug outside the United States and Canada, rights that Merck KGaA currently has with Erbitux.
At the time, ImClone stated that "commercial rights to this antibody in the U.S., Canada and Japan fall within the scope of ImClone Systems' commercial agreement with Bristol-Myers Squibb regarding Erbitux."
Officials from ImClone weren't immediately available to comment, but the company was under different management when the statement was issued.
Current management is said to frustrated with the 2006 statement, based on comments from a person familiar with the situation, but the company may try to justify that statement by claiming it refers to Bristol-Myers' right of first refusal for all of ImClone's pipeline products, which Bristol had until that right expired in September 2006.
ImClone also may claim that IMC-11F8 is actually a separate drug, not derived from Erbitux in any way, but that angle will force it to deal with the restrictions on developing a competing product that are contained in the agreement.
Under the agreement, a competing product is defined as one that "has as its only mechanism of action an antagonism of the EGF receptor."
Both drugs attack cancer cells by latching onto, and blocking, a growth- related trigger called epidermal growth-factor, or EGF, that occurs in some cancers.
"If this drug is therapeutically effective because it interferes with - or otherwise antagonizes or blocks or inhibits - the EGF receptor and does nothing else, then it is a competing product," said John P. Iwanicki, a patent attorney and senior partner specializing in the pharmaceutical industry with Banner & Witcoff Ltd in Boston.
If it is a competing product, it must either be divested or ImClone must offer Bristol-Myers to participate in the commercialization and development on a 50/50 basis.
Even then, ImClone still may have an argument. The company could argue that the restriction only applies to late-stage development in North America, where Bristol holds rights to Erbitux. IMC-11F8 is being developed in Europe, where ImClone controls all rights to the drug.
That argument may hold water, Iwanicki said, and ImClone could begin developing or commercializing IMC-11F8 in the U.S. after September 2008 when ban on competing products expires.
*************************
hier eine Meinung aus dem IMCL yahoo-Board:
Today I sold 5% of my imcl holdings. For 2 reasons, first I am way overbalanced in imcl and need to cash in, but second I’m afraid that there will be no buyout and erbitux will face stiff competition in the future. My fear is that since Imclone has never developed a product before, remember they bought erbitux, that they may never do it in the future. With Icahn as chair and JJ as CEO I am skeptical they can bring a product from the bench to the market. What drives imclone value is the expanded label, not the pipeline, IMO.
Take for example the competition for erbitux:
Imc-11F8 is a IgG1 fully human antibody isolated by ImClone using an antibody-fragment (Fab) phage display library licensed to the company by Dyax, and then further optimized by ImClone Systems.
Panitumumab is a IgG0 fully human antibody developed from the transgenic mice from mederax, it lacks the ADCC effect.
Zalutumumad is a IgG1,k fully human antibody developed from the medrax transgenic mice technique similar to panitumumab (k means its kappa light chain type, this makes it different from imc-11F8 which is a lambda light chain type, I think). Zalutumumad has the ADCC effect where pani did not. Zalutumumad is ahead of 11F8 in clinical studies and is fast tracked by the FDA for refractory H&N cancer. Zalu is not partnered with anybody and Genmab holds the worldwide rights. Read about it here.
Read about it here.
http://www.genmab.com/ScienceAndResearch...
EMD 7200 (Matuzumab) is a humanized IgG1 MAb against the EGFr. That failed in clinical trials and was being developed by eMerck.
Nimotuzumab from YMI is apparently a placebo.
The erbitux franchise could be lost with approval of zalu. Zalu could be better than 11F8.
Can 1121b compete against Avastin? Maybe, maybe not.
I think you get my point, that there is a lot of risk here that goes along with the possible very high reward.
As a significant shareholder (over 1M) I want imcl to negotiate the best deal possible and settle. Imclone cannot develop a world class biotech co with JJ and Carl in charge….IMHO
mfg ipollit
Beim Deal scheint auch der Erbitux-Nachfolger IMC-11F8 eine wesentliche Rolle zu spielen. IMC-11F8 ist allerdings erst in PII und deutlich hinter HuMax-EGFR. Daher ist es interessant, wie IMC-11F8 hier bewertet wird:
ImClone's Value May Rest On Debate Over Next Erbitux
August 07, 2008: 10:52 AM EST
NEW YORK -(Dow Jones)- The battle for ImClone Systems Inc. (IMCL) may come down to the fight over who has rights to the follow-up to the company's sole but increasingly successful product, the cancer treatment Erbitux.
ImClone claims to own the complete rights to the next-generation Erbitux, which carries high expectations but isn't seen on the market until well into the next decade. That interpretation contradicts the company's own statement two years ago and is disputed by Bristol-Myers Squibb Co. (BMY), which co-markets the current Erbitux in the U.S. and is seeking to buy the 83% of ImClone that it doesn't own for $4.5 billion.
ImClone has said Bristol's offer "greatly undervalues" the company partly because of its product pipeline, of which the next-generation Erbitux is a key part.
Resolving the next-generation Erbitux dispute could influence billions of dollars in sales in the next decade, ImClone's total value, the action of other possible suitors and whether ImClone attempts to spin off its pipeline, a move the company said it was considering.
The disagreement centers on whether the drug, called IMC-11F8, was derived in any way from the current Erbitux and if its development can be viewed as a " competing product," as defined by the 2001 agreement between the companies. If either of those are true, Bristol is entitled to rights to the drug.
"My read is that the contract language is nebulous," Cowen & Co. analyst Eric Schmidt said. There is "clearly room for (Bristol) to claim one thing and ( ImClone) the other."
Because of the confusion, the battle over IMC-11F8 could end up in front of an arbitrator and may take years to resolve - something that Bristol-Myers can avoid by buying ImClone, which may mean raising its offer of $60 a share. Wall Street expects a higher offer for ImClone as the stock closed Wednesday at $ 64.07 and recently traded at $64.09.
The IMC-11F8 dispute comes as the first-generation Erbitux gains traction. Erbitux is approved to treat head, neck and colorectal cancer; is expected to be filed for approval in lung cancer in the fourth quarter; and is being studied in multiple other forms of the disease.
UBS projects 2008 global Erbitux sales of $1.7 billion, up 31% from $1.3 billion in 2007, and sees that rising to $3.1 billion in 2011.
Such sales estimates aren't available for IMC-11F8, which won't enter pivotal trials - likely to last at least 2 1/2 years - until the first half of 2009. The success of the first-generation Erbitux reduces some risk on the follow-up because it has a similar target. The key difference between the two is that the new drug is a human antibody, expected to be safer and have less-frequent dosing, while Erbitux is a hybrid of a human and mouse antibody.
The unknown over IMC-11F8's rights is likely to give pause to other potential suitors of ImClone or any spinoff of its pipeline. That's because other companies may be less willing to pay billions for a company if they then have to share the profits on a key future drug.
Of course, if Bristol buys ImClone, that issue would be moot, which is why some might see the recent posturing - that started from ImClone before Bristol made its offer - as a bargaining tactic.
Conflicting Views
ImClone has asserted, as recently as two weeks ago, that it owns all the rights to IMC-11F8, and in a statement this week said that Bristol-Myers "may have no rights to market" the drug.
Bristol-Myers disagrees. "We believe we have the rights to 11F8 under our existing contractual agreement," a Bristol-Myers spokeswoman said this week, with no further comments on the company's strategy for claiming the rights.
The agreement, dating from 2001, covers Erbitux, along with "all fully humanized or human" versions, analogs or derivatives of the drug.
ImClone's recent comments even conflict with its own language from April 2006, when ImClone won an arbitration decision against Erbitux's international marketing partner, Germany's Merck KGaA (MRK.XE), for the international rights related to IMC-11F8. The decision gave ImClone the rights to develop and commercialize the drug outside the United States and Canada, rights that Merck KGaA currently has with Erbitux.
At the time, ImClone stated that "commercial rights to this antibody in the U.S., Canada and Japan fall within the scope of ImClone Systems' commercial agreement with Bristol-Myers Squibb regarding Erbitux."
Officials from ImClone weren't immediately available to comment, but the company was under different management when the statement was issued.
Current management is said to frustrated with the 2006 statement, based on comments from a person familiar with the situation, but the company may try to justify that statement by claiming it refers to Bristol-Myers' right of first refusal for all of ImClone's pipeline products, which Bristol had until that right expired in September 2006.
ImClone also may claim that IMC-11F8 is actually a separate drug, not derived from Erbitux in any way, but that angle will force it to deal with the restrictions on developing a competing product that are contained in the agreement.
Under the agreement, a competing product is defined as one that "has as its only mechanism of action an antagonism of the EGF receptor."
Both drugs attack cancer cells by latching onto, and blocking, a growth- related trigger called epidermal growth-factor, or EGF, that occurs in some cancers.
"If this drug is therapeutically effective because it interferes with - or otherwise antagonizes or blocks or inhibits - the EGF receptor and does nothing else, then it is a competing product," said John P. Iwanicki, a patent attorney and senior partner specializing in the pharmaceutical industry with Banner & Witcoff Ltd in Boston.
If it is a competing product, it must either be divested or ImClone must offer Bristol-Myers to participate in the commercialization and development on a 50/50 basis.
Even then, ImClone still may have an argument. The company could argue that the restriction only applies to late-stage development in North America, where Bristol holds rights to Erbitux. IMC-11F8 is being developed in Europe, where ImClone controls all rights to the drug.
That argument may hold water, Iwanicki said, and ImClone could begin developing or commercializing IMC-11F8 in the U.S. after September 2008 when ban on competing products expires.
*************************
hier eine Meinung aus dem IMCL yahoo-Board:
Today I sold 5% of my imcl holdings. For 2 reasons, first I am way overbalanced in imcl and need to cash in, but second I’m afraid that there will be no buyout and erbitux will face stiff competition in the future. My fear is that since Imclone has never developed a product before, remember they bought erbitux, that they may never do it in the future. With Icahn as chair and JJ as CEO I am skeptical they can bring a product from the bench to the market. What drives imclone value is the expanded label, not the pipeline, IMO.
Take for example the competition for erbitux:
Imc-11F8 is a IgG1 fully human antibody isolated by ImClone using an antibody-fragment (Fab) phage display library licensed to the company by Dyax, and then further optimized by ImClone Systems.
Panitumumab is a IgG0 fully human antibody developed from the transgenic mice from mederax, it lacks the ADCC effect.
Zalutumumad is a IgG1,k fully human antibody developed from the medrax transgenic mice technique similar to panitumumab (k means its kappa light chain type, this makes it different from imc-11F8 which is a lambda light chain type, I think). Zalutumumad has the ADCC effect where pani did not. Zalutumumad is ahead of 11F8 in clinical studies and is fast tracked by the FDA for refractory H&N cancer. Zalu is not partnered with anybody and Genmab holds the worldwide rights.
 Read about it here.
Read about it here. http://www.genmab.com/ScienceAndResearch...
EMD 7200 (Matuzumab) is a humanized IgG1 MAb against the EGFr. That failed in clinical trials and was being developed by eMerck.
Nimotuzumab from YMI is apparently a placebo.
The erbitux franchise could be lost with approval of zalu. Zalu could be better than 11F8.
Can 1121b compete against Avastin? Maybe, maybe not.
I think you get my point, that there is a lot of risk here that goes along with the possible very high reward.
As a significant shareholder (over 1M) I want imcl to negotiate the best deal possible and settle. Imclone cannot develop a world class biotech co with JJ and Carl in charge….IMHO
mfg ipollit
letzte Aktionen...
- Imclone entgültig liquidiert
- Regeneron weiter abgebaut
- Vertex halbiert
- neue Positionen in Exelixis und Array


mfg ipollit
- Imclone entgültig liquidiert
- Regeneron weiter abgebaut
- Vertex halbiert
- neue Positionen in Exelixis und Array
mfg ipollit
Antwort auf Beitrag Nr.: 34.719.369 von ipollit am 13.08.08 16:33:55damit sieht es aktuell so aus...
23,2% Genmab http://finance.yahoo.com/q?s=GEN.CO
9,6% Onyx http://finance.yahoo.com/q?s=ONXX
9,4% Regeneron http://finance.yahoo.com/q?s=REGN
6,7% OSI http://finance.yahoo.com/q?s=OSIP
6,5% Evotec http://finance.yahoo.com/q?s=EVT.DE
5,5% Intercell http://finance.yahoo.com/q?s=IJE.F
5,4% Cubist http://finance.yahoo.com/q?s=CBST
5,2% Isis http://finance.yahoo.com/q?s=ISIS
5,2% Exelixis http://finance.yahoo.com/q?s=EXEL
4,2% Array http://finance.yahoo.com/q?s=ARRY
3,7% Arena http://finance.yahoo.com/q?s=ARNA
3,2% ViroPharma http://finance.yahoo.com/q?s=VPHM
2,8% Micromet http://finance.yahoo.com/q?s=MITI
2,6% Incyte http://finance.yahoo.com/q?s=INCY
2,5% Rigel http://finance.yahoo.com/q?s=RIGL
2,5% Vertex http://finance.yahoo.com/q?s=VRTX
1,8% Supergen http://finance.yahoo.com/q?s=SUPG
Watchliste: POZN, ARIA, SGEN, MNTA, ITMN
mfg ipollit
23,2% Genmab http://finance.yahoo.com/q?s=GEN.CO
9,6% Onyx http://finance.yahoo.com/q?s=ONXX
9,4% Regeneron http://finance.yahoo.com/q?s=REGN
6,7% OSI http://finance.yahoo.com/q?s=OSIP
6,5% Evotec http://finance.yahoo.com/q?s=EVT.DE
5,5% Intercell http://finance.yahoo.com/q?s=IJE.F
5,4% Cubist http://finance.yahoo.com/q?s=CBST
5,2% Isis http://finance.yahoo.com/q?s=ISIS
5,2% Exelixis http://finance.yahoo.com/q?s=EXEL
4,2% Array http://finance.yahoo.com/q?s=ARRY
3,7% Arena http://finance.yahoo.com/q?s=ARNA
3,2% ViroPharma http://finance.yahoo.com/q?s=VPHM
2,8% Micromet http://finance.yahoo.com/q?s=MITI
2,6% Incyte http://finance.yahoo.com/q?s=INCY
2,5% Rigel http://finance.yahoo.com/q?s=RIGL
2,5% Vertex http://finance.yahoo.com/q?s=VRTX
1,8% Supergen http://finance.yahoo.com/q?s=SUPG
Watchliste: POZN, ARIA, SGEN, MNTA, ITMN
mfg ipollit
NBI

RT-Charts...




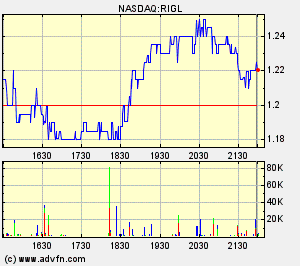


mfg ipollit
RT-Charts...
mfg ipollit
Hier versteht aber einer sein Handwerk!
Viel Glück mit deinen Positionen von meiner Seite, ich hoffe für dich, dass deine GPC-Kerbe bald wegpoliert werden kann!
Grüße!
Viel Glück mit deinen Positionen von meiner Seite, ich hoffe für dich, dass deine GPC-Kerbe bald wegpoliert werden kann!
Grüße!
NBI - neues Jahreshoch...


BTK - neues All-Time-High!


FUND VIEW-Fund firm OPM buys back into biotech sector
Thu Aug 14, 2008 9:10pm IST
LONDON, Aug 14 (Reuters) - Now is the right time to start investing in the biotechnology sector again, according to OPM Fund Management, which has recently bought into the Axa Framlington Biotech fund. OPM fund manager James Bruce believes the sector is likely to benefit as firms such as AstraZeneca (AZN.L: Quote, Profile, Research), Bristol-Myers Squibb (BMY.N: Quote, Profile, Research), GlaxoSmithkline (GLAX.BO: Quote, Profile, Research) and Pfizer (PFE.N: Quote, Profile, Research) face pressure on top-line growth from the expiration of drug patents and look to buy biotech firms for their drugs.
Whilst the biotech sector has been almost flat over the past five years, his comments come after a recent uptick in performance during a volatile period for markets overall.
Since the end of June the Nasdaq Biotechnology index has risen 15.5 percent, while the S&P 500 index .SPX is up just 0.5 percent. "The biotech industry continues to deliver many promising new drugs, making some of these companies an attractive proposition to the large pharmaceutical concerns," Bruce said in a note.
"In a drive to replenish their drug portfolios, it is no surprise to see the majors digging deep in to their pockets to acquire such companies and their pipeline of drugs."
Biotechnology has seen a wave of takeover activity in the last month.
In the United States, Roche (ROG.VX: Quote, Profile, Research) is offering $43.7 billion for the rest of Genentech (DNA.N: Quote, Profile, Research) it does not already own and Bristol-Myers Squibb has bid $5.2 billion for ImClone Systems (IMCL.O: Quote, Profile, Research).
On a smaller scale, Sanofi-Aventis (SASY.PA: Quote, Profile, Research) agreed last month to buy British vaccine firm Acambis (ACM.L: Quote, Profile, Research) for around $550 million, or a 64 percent premium to the prevailing share price.
OPM has recently bought into Axa Framlington Biotech fund which is up 2 percent over the past three years, compared with a 5.5 percent gain in the Nasdaq Biotechnology index. Since April it has been run by new manager Andy Smith. On Wednesday venture capital firm Index Ventures said bombed-out valuations in the European biotech sector presented an unprecedented buying opportunity. (Reporting by Laurence Fletcher; editing by Tony Austin)
mal sehen wie weit das ganze gehen wird... vielleicht laufen wir bald auch schon in das abschließende Hoch hinein. Einen größeren Teil der Strecke haben wir ja bereits zurückgelegt.
mfg ipollit
BTK - neues All-Time-High!
FUND VIEW-Fund firm OPM buys back into biotech sector
Thu Aug 14, 2008 9:10pm IST
LONDON, Aug 14 (Reuters) - Now is the right time to start investing in the biotechnology sector again, according to OPM Fund Management, which has recently bought into the Axa Framlington Biotech fund. OPM fund manager James Bruce believes the sector is likely to benefit as firms such as AstraZeneca (AZN.L: Quote, Profile, Research), Bristol-Myers Squibb (BMY.N: Quote, Profile, Research), GlaxoSmithkline (GLAX.BO: Quote, Profile, Research) and Pfizer (PFE.N: Quote, Profile, Research) face pressure on top-line growth from the expiration of drug patents and look to buy biotech firms for their drugs.
Whilst the biotech sector has been almost flat over the past five years, his comments come after a recent uptick in performance during a volatile period for markets overall.
Since the end of June the Nasdaq Biotechnology index has risen 15.5 percent, while the S&P 500 index .SPX is up just 0.5 percent. "The biotech industry continues to deliver many promising new drugs, making some of these companies an attractive proposition to the large pharmaceutical concerns," Bruce said in a note.
"In a drive to replenish their drug portfolios, it is no surprise to see the majors digging deep in to their pockets to acquire such companies and their pipeline of drugs."
Biotechnology has seen a wave of takeover activity in the last month.
In the United States, Roche (ROG.VX: Quote, Profile, Research) is offering $43.7 billion for the rest of Genentech (DNA.N: Quote, Profile, Research) it does not already own and Bristol-Myers Squibb has bid $5.2 billion for ImClone Systems (IMCL.O: Quote, Profile, Research).
On a smaller scale, Sanofi-Aventis (SASY.PA: Quote, Profile, Research) agreed last month to buy British vaccine firm Acambis (ACM.L: Quote, Profile, Research) for around $550 million, or a 64 percent premium to the prevailing share price.
OPM has recently bought into Axa Framlington Biotech fund which is up 2 percent over the past three years, compared with a 5.5 percent gain in the Nasdaq Biotechnology index. Since April it has been run by new manager Andy Smith. On Wednesday venture capital firm Index Ventures said bombed-out valuations in the European biotech sector presented an unprecedented buying opportunity. (Reporting by Laurence Fletcher; editing by Tony Austin)
mal sehen wie weit das ganze gehen wird... vielleicht laufen wir bald auch schon in das abschließende Hoch hinein. Einen größeren Teil der Strecke haben wir ja bereits zurückgelegt.
mfg ipollit
Antwort auf Beitrag Nr.: 34.730.902 von ipollit am 14.08.08 20:37:13einen schönen Chart-Überblick aller im NBI enthaltenen Biotechs...
http://www.ftor.de/board/showthread.php?t=24245&page=1
mfg ipollit
http://www.ftor.de/board/showthread.php?t=24245&page=1
mfg ipollit
Hast ja glaub ich auch im Depot
14.08.2008 20:01:00 (HUGIN)
Clinical Data Published in Science Show Tumor Regressions in Relapsed Lymphoma Patients Treated with T Cell Engaging BiTE Antibody Blinatumomab
Corporate news announcement processed and transmitted by Hugin ASA. The issuer is solely responsible for the content of this announcement. ---------------------------------------------------------------------- -------------- First T cell engaging antibody (BiTE) showing clinical benefit in cancer patients; Blinatumomab enables patients own T cells to recognize and attack cancer cells BETHESDA, MD - August 14, 2008 -- Micromet, Inc. (Nasdaq: MITI), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced publication of a Phase 1 clinical study[1] on its BiTE® antibody blinatumomab (MT103/MEDI-538) in this week's issue of Science. The article is available at www.sciencemag.org. Blinatumomab is being co-developed with MedImmune. Blinatumomab is a novel antibody therapy that activates a patient's T-cells to seek out and destroy cancer cells. The phase 1 study demonstrated tumor regression, and in some cases, complete remission, in non-Hodgkin's lymphoma patients who relapsed after previous treatments and were considered to have incurable disease. Most of the remissions are reported to continue, with the longest remission ongoing for more than one year. Results from this ongoing Phase 1 clinical trial with the CD19-specific BiTE antibody blinatumomab show that all seven patients treated to date at 0.06 mg/m2 per day achieved complete or partial responses. The safety profile observed in this study supports continued blinatumomab development. "These results represent significant progress of a T cell engaging antibody for treatment of lymphoma patients as single agent therapy. We observed tumor regression in patients at serum levels of blinatumomab, which are approximately five orders of magnitude lower than serum levels needed by conventional monoclonal antibodies for achieving a tumor regression in this disease. This may relate to the high anti-tumor activity of cytotoxic T cells recruited by blinatumomab," commented Micromet Senior Vice President and Chief Scientific Officer Patrick Baeuerle. "This first observation of durable objective responses in relapsed, incurable patients indicates the potential blinatumomab and BiTE antibodies in general may have in fighting cancer," added Micromet Senior Vice President and Chief Medical Officer Carsten Reinhardt, M.D. (more) Typically antibodies cannot engage T cells because T cells lack the appropriate receptors for binding antibodies. Previous attempts have shown the potential of T cells to treat cancer, but the therapeutic approaches tested to date have been hampered by cancer cells' ability to escape recognition by T cells. The use of antibodies that are specifically designed to engage T cells for attacking cancer cells may provide a more effective anti-tumor approach than conventional monoclonal antibodies, which require much higher doses and are typically combined with chemotherapies. Micromet has additional clinical trials with BiTE antibodies underway, including a phase 2 clinical trial to evaluate blinatumomab for the treatment of patients with acute lymphoblastic leukemia (ALL), and a phase 1 trial investigating MT110, a BiTE antibody targeting EpCAM, in patients with lung or gastrointestinal cancers. Micromet will host a webcast and a conference call on Monday, August 18 at 10:00 a.m. Eastern Time , (4:00 p.m. Central European Time), to discuss these results. The webcast can be accessed at: www.micromet-inc.com/sciencepub. To participate in the conference call, dial 866-202-4367 (U.S.) or 617-213-8845 (international), passcode: 31176615. [1]Bargou R et al. (2008) Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321: 974-977 (2008) About Science Founded in 1880 on $10,000 of seed money from the American inventor Thomas Edison, Science has grown to become the world's leading outlet for scientific news, commentary and cutting-edge research, with the largest paid circulation of any peer-reviewed general-science journal. Through its print and online incarnations, Science reaches an estimated worldwide readership of more than one million. In content, too, the journal is truly international in scope; some 35 to 40 percent of the corresponding authors on its papers are based outside the United States. Its articles consistently rank among world's most cited research. About BiTE Antibodies BiTE® antibodies are designed to direct the body's cytotoxic, or cell- destroying, T cells against tumor cells, and represent a new therapeutic approach to cancer therapy. BiTE antibodies have been shown to induce an immunological synapse between a T cell and a (more) tumor cell in the same manner as observed during physiological T cell attacks. These cytolytic synapses enable the delivery of cytotoxic proteins from T cells into tumor cells, ultimately inducing a self-destruction process in the tumor cell referred to as apoptosis, or programmed cell death. In the presence of BiTE antibodies, T cells have been demonstrated to serially eliminate tumor cells, which explains the activity of BiTE antibodies at very low concentrations and at very low ratios of T cells to target cells. Through the process of killing cancer cells, T cells proliferate, which leads to an increased number of T cells at the site of attack. Several antibodies in Micromet's product pipeline are BiTE antibodies and have been generated based on Micromet's proprietary BiTE antibody platform. The most advanced BiTE antibody is blinatumomab (MT103/MEDI-538), targeting CD19, and has provided proof-of-concept in an ongoing phase 1 clinical trial in patients with advanced non-Hodgkin's lymphoma. MT110, which is targeting EpCAM (CD326) and is the first BiTE antibody with potential applications in the treatment of solid tumors, is in a phase 1 clinical trial in patients with lung or gastrointestinal cancers. Three additional BiTE antibodies, targeting CD33, CEA and MCSP, respectively, are in preclinical development. About Micromet, Inc. Micromet, Inc. (www.micromet-inc.com) is a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases. Four of its antibodies are currently in clinical trials, while the remainder of the product pipeline is in preclinical development. The BiTE® antibody blinatumomab (MT103/MEDI-538) is in a phase 2 clinical trial for the treatment of patients with acute lymphoblastic leukemia and in a phase 1 clinical trial for the treatment of patients with non-Hodgkin's lymphoma. BiTE antibodies represent a new class of antibodies that activate a patient's own cytotoxic T cells, considered the most powerful "killer cells" of the human immune system, to eliminate cancer cells. Micromet is developing blinatumomab in collaboration with MedImmune, Inc., a subsidiary of AstraZeneca plc. MT110 is the second BiTE antibody in clinical trials, and is being developed by Micromet in a phase 1 clinical trial for the treatment of patients with lung or gastrointestinal cancer. The third clinical stage antibody is adecatumumab, also known as MT201, a human monoclonal antibody that targets epithelial cell adhesion molecule (EpCAM)-expressing solid tumors. Micromet is developing adecatumumab in collaboration with Merck Serono in a phase 1b clinical trial evaluating adecatumumab in combination with docetaxel for the treatment of patients with metastatic breast cancer. The fourth clinical (more) stage antibody is MT293 which is licensed to TRACON Pharmaceuticals, Inc. and is being developed in a phase 1 clinical trial for the treatment of patients with cancer. Three additional BiTE antibodies, targeting CD33, CEA and MCSP, respectively, are in preclinical development. In addition, Micromet has established a collaboration with Nycomed for the development and commercialization of MT203, a human antibody neutralizing the activity of granulocyte/macrophage colony stimulating factor (GM-CSF), which has potential applications in the treatment of various inflammatory and autoimmune diseases, such as rheumatoid arthritis, psoriasis, or multiple sclerosis. Forward-Looking Statements This release contains certain forward-looking statements that involve risks and uncertainties that could cause actual results to be materially different from historical results or from any future results expressed or implied by such forward-looking statements. These forward-looking statements include statements regarding the efficacy, safety and intended utilization of our product candidates, the development of our BiTE antibody technology, the conduct, timing and results of future clinical trials, expectations of the future expansion of our product pipeline and collaborations, and our plans regarding future presentations of clinical data. You are urged to consider statements that include the words "ongoing," "may," "will," "believes," "potential," "expects," "plans," "anticipates," "intends," or the negative of those words or other similar words to be uncertain and forward-looking. Factors that may cause actual results to differ materially from any future results expressed or implied by any forward-looking statements include the risk that product candidates that appeared promising in early research, preclinical studies or clinical trials do not demonstrate safety and/or efficacy in subsequent clinical trials, the risk that encouraging results from early research, preclinical studies or clinical trials may not be confirmed upon further analysis of the detailed results of such research, preclinical study or clinical trial, the risk that additional information relating to the safety, efficacy or tolerability of our product candidates may be discovered upon further analysis of preclinical or clinical trial data, the risk that we or our collaborators will not obtain approval to market our product candidates, the risks associated with reliance on outside financing to meet capital requirements, and the risks associated with reliance on collaborators, including MedImmune, Merck Serono, TRACON and Nycomed, for the funding or conduct of further development and commercialization activities relating to our product candidates. These factors and others are more fully discussed in Micromet's Annual Report on Form 10-K for the fiscal year ended December 31, 2007, filed with the SEC on March 14, 2008, as well as other filings by the company with the SEC. (more) Any forward-looking statements are made pursuant to Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, and, as such, speak only as of the date made. Micromet, Inc. undertakes no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise. # # # Contact Information US Media: European Media: Andrea tenBroek/Chris Stamm Ludger Wess (781)-684-0770 +49 (40) 8816 5964 micromet@schwartz-pr.com ludger@akampion.com US Investors: European Investors: Susan Noonan Ines-Regina Buth (212) 966-3650 +49 (30) 2363 2768 susan@sanoonan.com ines@akampion.com --- End of Message --- Micromet Inc. 2110 Rutherford Road Carlsbad USA WKN: A0JMQD; ISIN: US59509C1053; Listed: Xetra Stars in Frankfurter Wertpapierbörse; http://www.micromet.de Copyright © Hugin AS 2008. All rights reserved.
14.08.2008 20:01:00 (HUGIN)
Clinical Data Published in Science Show Tumor Regressions in Relapsed Lymphoma Patients Treated with T Cell Engaging BiTE Antibody Blinatumomab
Corporate news announcement processed and transmitted by Hugin ASA. The issuer is solely responsible for the content of this announcement. ---------------------------------------------------------------------- -------------- First T cell engaging antibody (BiTE) showing clinical benefit in cancer patients; Blinatumomab enables patients own T cells to recognize and attack cancer cells BETHESDA, MD - August 14, 2008 -- Micromet, Inc. (Nasdaq: MITI), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced publication of a Phase 1 clinical study[1] on its BiTE® antibody blinatumomab (MT103/MEDI-538) in this week's issue of Science. The article is available at www.sciencemag.org. Blinatumomab is being co-developed with MedImmune. Blinatumomab is a novel antibody therapy that activates a patient's T-cells to seek out and destroy cancer cells. The phase 1 study demonstrated tumor regression, and in some cases, complete remission, in non-Hodgkin's lymphoma patients who relapsed after previous treatments and were considered to have incurable disease. Most of the remissions are reported to continue, with the longest remission ongoing for more than one year. Results from this ongoing Phase 1 clinical trial with the CD19-specific BiTE antibody blinatumomab show that all seven patients treated to date at 0.06 mg/m2 per day achieved complete or partial responses. The safety profile observed in this study supports continued blinatumomab development. "These results represent significant progress of a T cell engaging antibody for treatment of lymphoma patients as single agent therapy. We observed tumor regression in patients at serum levels of blinatumomab, which are approximately five orders of magnitude lower than serum levels needed by conventional monoclonal antibodies for achieving a tumor regression in this disease. This may relate to the high anti-tumor activity of cytotoxic T cells recruited by blinatumomab," commented Micromet Senior Vice President and Chief Scientific Officer Patrick Baeuerle. "This first observation of durable objective responses in relapsed, incurable patients indicates the potential blinatumomab and BiTE antibodies in general may have in fighting cancer," added Micromet Senior Vice President and Chief Medical Officer Carsten Reinhardt, M.D. (more) Typically antibodies cannot engage T cells because T cells lack the appropriate receptors for binding antibodies. Previous attempts have shown the potential of T cells to treat cancer, but the therapeutic approaches tested to date have been hampered by cancer cells' ability to escape recognition by T cells. The use of antibodies that are specifically designed to engage T cells for attacking cancer cells may provide a more effective anti-tumor approach than conventional monoclonal antibodies, which require much higher doses and are typically combined with chemotherapies. Micromet has additional clinical trials with BiTE antibodies underway, including a phase 2 clinical trial to evaluate blinatumomab for the treatment of patients with acute lymphoblastic leukemia (ALL), and a phase 1 trial investigating MT110, a BiTE antibody targeting EpCAM, in patients with lung or gastrointestinal cancers. Micromet will host a webcast and a conference call on Monday, August 18 at 10:00 a.m. Eastern Time , (4:00 p.m. Central European Time), to discuss these results. The webcast can be accessed at: www.micromet-inc.com/sciencepub. To participate in the conference call, dial 866-202-4367 (U.S.) or 617-213-8845 (international), passcode: 31176615. [1]Bargou R et al. (2008) Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321: 974-977 (2008) About Science Founded in 1880 on $10,000 of seed money from the American inventor Thomas Edison, Science has grown to become the world's leading outlet for scientific news, commentary and cutting-edge research, with the largest paid circulation of any peer-reviewed general-science journal. Through its print and online incarnations, Science reaches an estimated worldwide readership of more than one million. In content, too, the journal is truly international in scope; some 35 to 40 percent of the corresponding authors on its papers are based outside the United States. Its articles consistently rank among world's most cited research. About BiTE Antibodies BiTE® antibodies are designed to direct the body's cytotoxic, or cell- destroying, T cells against tumor cells, and represent a new therapeutic approach to cancer therapy. BiTE antibodies have been shown to induce an immunological synapse between a T cell and a (more) tumor cell in the same manner as observed during physiological T cell attacks. These cytolytic synapses enable the delivery of cytotoxic proteins from T cells into tumor cells, ultimately inducing a self-destruction process in the tumor cell referred to as apoptosis, or programmed cell death. In the presence of BiTE antibodies, T cells have been demonstrated to serially eliminate tumor cells, which explains the activity of BiTE antibodies at very low concentrations and at very low ratios of T cells to target cells. Through the process of killing cancer cells, T cells proliferate, which leads to an increased number of T cells at the site of attack. Several antibodies in Micromet's product pipeline are BiTE antibodies and have been generated based on Micromet's proprietary BiTE antibody platform. The most advanced BiTE antibody is blinatumomab (MT103/MEDI-538), targeting CD19, and has provided proof-of-concept in an ongoing phase 1 clinical trial in patients with advanced non-Hodgkin's lymphoma. MT110, which is targeting EpCAM (CD326) and is the first BiTE antibody with potential applications in the treatment of solid tumors, is in a phase 1 clinical trial in patients with lung or gastrointestinal cancers. Three additional BiTE antibodies, targeting CD33, CEA and MCSP, respectively, are in preclinical development. About Micromet, Inc. Micromet, Inc. (www.micromet-inc.com) is a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases. Four of its antibodies are currently in clinical trials, while the remainder of the product pipeline is in preclinical development. The BiTE® antibody blinatumomab (MT103/MEDI-538) is in a phase 2 clinical trial for the treatment of patients with acute lymphoblastic leukemia and in a phase 1 clinical trial for the treatment of patients with non-Hodgkin's lymphoma. BiTE antibodies represent a new class of antibodies that activate a patient's own cytotoxic T cells, considered the most powerful "killer cells" of the human immune system, to eliminate cancer cells. Micromet is developing blinatumomab in collaboration with MedImmune, Inc., a subsidiary of AstraZeneca plc. MT110 is the second BiTE antibody in clinical trials, and is being developed by Micromet in a phase 1 clinical trial for the treatment of patients with lung or gastrointestinal cancer. The third clinical stage antibody is adecatumumab, also known as MT201, a human monoclonal antibody that targets epithelial cell adhesion molecule (EpCAM)-expressing solid tumors. Micromet is developing adecatumumab in collaboration with Merck Serono in a phase 1b clinical trial evaluating adecatumumab in combination with docetaxel for the treatment of patients with metastatic breast cancer. The fourth clinical (more) stage antibody is MT293 which is licensed to TRACON Pharmaceuticals, Inc. and is being developed in a phase 1 clinical trial for the treatment of patients with cancer. Three additional BiTE antibodies, targeting CD33, CEA and MCSP, respectively, are in preclinical development. In addition, Micromet has established a collaboration with Nycomed for the development and commercialization of MT203, a human antibody neutralizing the activity of granulocyte/macrophage colony stimulating factor (GM-CSF), which has potential applications in the treatment of various inflammatory and autoimmune diseases, such as rheumatoid arthritis, psoriasis, or multiple sclerosis. Forward-Looking Statements This release contains certain forward-looking statements that involve risks and uncertainties that could cause actual results to be materially different from historical results or from any future results expressed or implied by such forward-looking statements. These forward-looking statements include statements regarding the efficacy, safety and intended utilization of our product candidates, the development of our BiTE antibody technology, the conduct, timing and results of future clinical trials, expectations of the future expansion of our product pipeline and collaborations, and our plans regarding future presentations of clinical data. You are urged to consider statements that include the words "ongoing," "may," "will," "believes," "potential," "expects," "plans," "anticipates," "intends," or the negative of those words or other similar words to be uncertain and forward-looking. Factors that may cause actual results to differ materially from any future results expressed or implied by any forward-looking statements include the risk that product candidates that appeared promising in early research, preclinical studies or clinical trials do not demonstrate safety and/or efficacy in subsequent clinical trials, the risk that encouraging results from early research, preclinical studies or clinical trials may not be confirmed upon further analysis of the detailed results of such research, preclinical study or clinical trial, the risk that additional information relating to the safety, efficacy or tolerability of our product candidates may be discovered upon further analysis of preclinical or clinical trial data, the risk that we or our collaborators will not obtain approval to market our product candidates, the risks associated with reliance on outside financing to meet capital requirements, and the risks associated with reliance on collaborators, including MedImmune, Merck Serono, TRACON and Nycomed, for the funding or conduct of further development and commercialization activities relating to our product candidates. These factors and others are more fully discussed in Micromet's Annual Report on Form 10-K for the fiscal year ended December 31, 2007, filed with the SEC on March 14, 2008, as well as other filings by the company with the SEC. (more) Any forward-looking statements are made pursuant to Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, and, as such, speak only as of the date made. Micromet, Inc. undertakes no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise. # # # Contact Information US Media: European Media: Andrea tenBroek/Chris Stamm Ludger Wess (781)-684-0770 +49 (40) 8816 5964 micromet@schwartz-pr.com ludger@akampion.com US Investors: European Investors: Susan Noonan Ines-Regina Buth (212) 966-3650 +49 (30) 2363 2768 susan@sanoonan.com ines@akampion.com --- End of Message --- Micromet Inc. 2110 Rutherford Road Carlsbad USA WKN: A0JMQD; ISIN: US59509C1053; Listed: Xetra Stars in Frankfurter Wertpapierbörse; http://www.micromet.de Copyright © Hugin AS 2008. All rights reserved.
Antwort auf Beitrag Nr.: 34.731.268 von schnappi am 14.08.08 21:26:03Micromet... ja, danke - die Technolgie scheint vielversprechend zu sein. Ist ja ein deutsches Unternehmen, was sich über den Einkauf eines Amis das US-Listing verschafft hat. Allerdings ist der Cash-Vorrat ein wenig bedenklich.
(wohl etwas populär...)
Donnerstag, 14. August 2008
Antikörper soll Blutkrebs heilen
Hoffnung für Krebspatienten
Deutsche Wissenschaftler haben eine neue Blutkrebs-Therapie aus einem gentechnisch hergestellten Antikörper entwickelt. Der neue Antikörper bringe das körpereigene Immunsystem dazu, den Krebs zu bekämpfen, schreiben Forscher um Ralf Bargou von der Universität Würzburg im US-Journal "Science".
Bei einer ersten Studie mit 38 Patienten mit der Blutkrebsart Non- Hodgkin-Lymphom hätten 11 angesprochen, erläuterte Patrick Baeuerle von der in München und den USA ansässigen Biotech-Firma Micromet. Sie hat den Antikörper entwickelt und zusammen mit den Universitätskliniken München, Würzburg, Essen, Ulm und Mainz erstmals an Patienten erprobt.
Hohe Erfolgsaussichten, gute Verträglichkeit
Bei der höchsten Dosierung, die bei sieben unheilbar kranken Patienten getestet wurde, habe der Antikörper sogar in allen Fällen angesprochen. "Das ist sehr ungewöhnlich", sagte Baeuerle. Von einer Heilung könne man aber noch nicht sprechen.
Die Therapie sei bei den bisherigen Dosierungen insgesamt gut verträglich geblieben. "Ganz sicher sehen wir nicht die klassischen Nebenwirkungen einer Chemotherapie", sagte Baeuerle. "Die meisten Patienten bekommen bei der Behandlung Fieber und Schüttelfrost wie beim Beginn einer Grippe. Das lässt aber nach ein paar Tagen nach." Auch die meisten anderen Nebenwirkungen seien meist von begrenzter Dauer und insgesamt gut beherrschbar gewesen.
Antik örper mit Brille
Der neue Antikörper namens Blinatumomab hat laut Baeuerle zwei Greifarme. Mit dem einen hakt er sich an den Tumorzellen fest, mit dem anderen fängt er sogenannte T-Zellen, die im Körper praktisch als "Polizei" entartete Zellen bekämpfen sollen. Bei Krebs hätten diese Killer-T-Zellen die Fähigkeit verloren, den Tumor zu erkennen. Deshalb könne das Immunsystem den Tumor nicht bekämpfen. "Was wir tun, ist, dass wir diesen T-Zellen wieder eine Brille aufsetzen." Indem die Antikörper die T-Zellen nahe an den Tumor heranführten, würden diese wieder aktiviert, gegen den Krebs vorzugehen. "Wir heilen uns also selbst mit unseren eigenen Immunzellen."
Seit Dezember laufe auch eine Studie mit Patienten mit akuter aggressiver Leukämie, dort könnten aber noch keine Ergebnisse veröffentlicht werden. Eine weitere Studie mit einem neuen Antikörper, der T-Zellen einfängt, habe bei Patienten mit Magen-, Darm- und Lungenkrebs begonnen.
mfg ipollit
(wohl etwas populär...)
Donnerstag, 14. August 2008
Antikörper soll Blutkrebs heilen
Hoffnung für Krebspatienten
Deutsche Wissenschaftler haben eine neue Blutkrebs-Therapie aus einem gentechnisch hergestellten Antikörper entwickelt. Der neue Antikörper bringe das körpereigene Immunsystem dazu, den Krebs zu bekämpfen, schreiben Forscher um Ralf Bargou von der Universität Würzburg im US-Journal "Science".
Bei einer ersten Studie mit 38 Patienten mit der Blutkrebsart Non- Hodgkin-Lymphom hätten 11 angesprochen, erläuterte Patrick Baeuerle von der in München und den USA ansässigen Biotech-Firma Micromet. Sie hat den Antikörper entwickelt und zusammen mit den Universitätskliniken München, Würzburg, Essen, Ulm und Mainz erstmals an Patienten erprobt.
Hohe Erfolgsaussichten, gute Verträglichkeit
Bei der höchsten Dosierung, die bei sieben unheilbar kranken Patienten getestet wurde, habe der Antikörper sogar in allen Fällen angesprochen. "Das ist sehr ungewöhnlich", sagte Baeuerle. Von einer Heilung könne man aber noch nicht sprechen.
Die Therapie sei bei den bisherigen Dosierungen insgesamt gut verträglich geblieben. "Ganz sicher sehen wir nicht die klassischen Nebenwirkungen einer Chemotherapie", sagte Baeuerle. "Die meisten Patienten bekommen bei der Behandlung Fieber und Schüttelfrost wie beim Beginn einer Grippe. Das lässt aber nach ein paar Tagen nach." Auch die meisten anderen Nebenwirkungen seien meist von begrenzter Dauer und insgesamt gut beherrschbar gewesen.
Antik örper mit Brille
Der neue Antikörper namens Blinatumomab hat laut Baeuerle zwei Greifarme. Mit dem einen hakt er sich an den Tumorzellen fest, mit dem anderen fängt er sogenannte T-Zellen, die im Körper praktisch als "Polizei" entartete Zellen bekämpfen sollen. Bei Krebs hätten diese Killer-T-Zellen die Fähigkeit verloren, den Tumor zu erkennen. Deshalb könne das Immunsystem den Tumor nicht bekämpfen. "Was wir tun, ist, dass wir diesen T-Zellen wieder eine Brille aufsetzen." Indem die Antikörper die T-Zellen nahe an den Tumor heranführten, würden diese wieder aktiviert, gegen den Krebs vorzugehen. "Wir heilen uns also selbst mit unseren eigenen Immunzellen."
Seit Dezember laufe auch eine Studie mit Patienten mit akuter aggressiver Leukämie, dort könnten aber noch keine Ergebnisse veröffentlicht werden. Eine weitere Studie mit einem neuen Antikörper, der T-Zellen einfängt, habe bei Patienten mit Magen-, Darm- und Lungenkrebs begonnen.
mfg ipollit
zu ISIS (nicht ganz aktuell)
http://www.isispharm.com/index.html
Strong Isis Pharma Outlook
Wednesday June 25, 1:38 pm ET
By Jason Napodano, CFA
We are reiterating our Buy rating on Isis Pharmaceuticals Inc. (NasdaqGM: ISIS - News). We believe that antisense technology represents an exciting and potentially revolutionary platform for developing therapeutic candidates to treat a wide variety of diseases. We see Isis as uniquely positioned for a very successful future.
The company's lead candidates are mipomersen for high cholesterol and ISIS-113715 for diabetes, along with several other proprietary and partnered programs for oncology, inflammatory disease, asthma, and viral infections. We think that by 2011 two to three additional antisense drugs developed by Isis could be on the market. Our financial model forecasts profitability in 2011.
We find it comforting that Genzyme (NasdaqGS: GENZ - News) recently agreed to acquire five million shares of Isis at $30 per share - $10 above our near-term target. We also encourage investors to pick up the shares at this level considering the drop in the stock price after what we believe was a significant over-reaction to the downside on the mipomersen news.
Thanks to collaborations, Isis has put itself in constant position to receive developmental milestone. The company sees no need to re-tap the financial markets in the next five years. The management expects to exit 2008 with over $450 million still on the books very conservative guidance in our view. If we couple the cash with the potential of mipomersen as a blockbuster and consider that several other compounds are moving nicely forward, Isis looks like the premier small-to-mid cap biotechnology company in our universe.
********
Isis Pharmaceuticals' Obesity Buster an Enormous Finding
by: Steven Turner posted on: June 27, 2008 | about stocks: ISIS
Logic may dictate that Isis Pharmaceuticals' largest multibillion-dollar blockbuster drug is its anti-lipid drug Mipomersen. After all, Genzyme (GENZ) has already paid Isis 325 million dollars upfront, and is scheduled to pay an additional 1.575 billion dollars in milestone payments.
True, Mipomersen has already proven to reverse atherosclerotic plaques in the murine model at unprecedented efficacy (up to 92%). It has also been able to lower lipid levels never achieved with traditional medicines in patients afflicted with homozygous hyperlipidemea.
I would even venture to say when fully approved, it would beat Pfizer's (PFE) Lipitor in gross annual sales and profits. So shouldn't Isis' Mipomersen be its largest medicinal candidate in terms of gross revenues and profitability potential?
No, not at all. While Mipomersen will turn out to be wildly successful, Isis' hidden gem is its obesity-busting diabetic drug ISIS 113715, a second-generation antisense inhibitor of protein tyrosine phosphatase 1b. It is this enzyme that inhibits insulin receptors in type 2 diabetics. This is no small feat, as there are over twenty-two million Americans suffering from this disease.
Even more important, a whopping 63% of Americans, or roughly 191 million people, according to the CDC, are overweight. A third of those are obese. And it is this drug that normalizes glucose but does not cause hypoglycemia like some oral diabetic agents. It also decreases circulating lipids to the point of decreasing the amount of fat in an over satiated (overfed) individual.
This effect has been noted in several scientific publications discussing inhibiting tyrosine phosphatase 1b. The problem was finding a specific antisense molecule that inhibited this individual phosphatase and not the general class of 1b phosphatases. Isis owns that particular antisense molecule.
I cannot overemphasize the importance of this finding. This medicine could be America's answer to its obesity epidemic.

mfg ipollit
http://www.isispharm.com/index.html
Strong Isis Pharma Outlook
Wednesday June 25, 1:38 pm ET
By Jason Napodano, CFA
We are reiterating our Buy rating on Isis Pharmaceuticals Inc. (NasdaqGM: ISIS - News). We believe that antisense technology represents an exciting and potentially revolutionary platform for developing therapeutic candidates to treat a wide variety of diseases. We see Isis as uniquely positioned for a very successful future.
The company's lead candidates are mipomersen for high cholesterol and ISIS-113715 for diabetes, along with several other proprietary and partnered programs for oncology, inflammatory disease, asthma, and viral infections. We think that by 2011 two to three additional antisense drugs developed by Isis could be on the market. Our financial model forecasts profitability in 2011.
We find it comforting that Genzyme (NasdaqGS: GENZ - News) recently agreed to acquire five million shares of Isis at $30 per share - $10 above our near-term target. We also encourage investors to pick up the shares at this level considering the drop in the stock price after what we believe was a significant over-reaction to the downside on the mipomersen news.
Thanks to collaborations, Isis has put itself in constant position to receive developmental milestone. The company sees no need to re-tap the financial markets in the next five years. The management expects to exit 2008 with over $450 million still on the books very conservative guidance in our view. If we couple the cash with the potential of mipomersen as a blockbuster and consider that several other compounds are moving nicely forward, Isis looks like the premier small-to-mid cap biotechnology company in our universe.
********
Isis Pharmaceuticals' Obesity Buster an Enormous Finding
by: Steven Turner posted on: June 27, 2008 | about stocks: ISIS
Logic may dictate that Isis Pharmaceuticals' largest multibillion-dollar blockbuster drug is its anti-lipid drug Mipomersen. After all, Genzyme (GENZ) has already paid Isis 325 million dollars upfront, and is scheduled to pay an additional 1.575 billion dollars in milestone payments.
True, Mipomersen has already proven to reverse atherosclerotic plaques in the murine model at unprecedented efficacy (up to 92%). It has also been able to lower lipid levels never achieved with traditional medicines in patients afflicted with homozygous hyperlipidemea.
I would even venture to say when fully approved, it would beat Pfizer's (PFE) Lipitor in gross annual sales and profits. So shouldn't Isis' Mipomersen be its largest medicinal candidate in terms of gross revenues and profitability potential?
No, not at all. While Mipomersen will turn out to be wildly successful, Isis' hidden gem is its obesity-busting diabetic drug ISIS 113715, a second-generation antisense inhibitor of protein tyrosine phosphatase 1b. It is this enzyme that inhibits insulin receptors in type 2 diabetics. This is no small feat, as there are over twenty-two million Americans suffering from this disease.
Even more important, a whopping 63% of Americans, or roughly 191 million people, according to the CDC, are overweight. A third of those are obese. And it is this drug that normalizes glucose but does not cause hypoglycemia like some oral diabetic agents. It also decreases circulating lipids to the point of decreasing the amount of fat in an over satiated (overfed) individual.
This effect has been noted in several scientific publications discussing inhibiting tyrosine phosphatase 1b. The problem was finding a specific antisense molecule that inhibited this individual phosphatase and not the general class of 1b phosphatases. Isis owns that particular antisense molecule.
I cannot overemphasize the importance of this finding. This medicine could be America's answer to its obesity epidemic.
mfg ipollit
Antwort auf Beitrag Nr.: 34.731.424 von ipollit am 14.08.08 21:45:15ein ältere paar Artikel zu Micromet...
Micromet, Inc.: The Real Star of ASH 2007
by: Ohad Hammer posted on: December 14, 2007 | about stocks: MITI
A lot of clinical data was published at the American Society of Hematology [ASH] meeting, some of it quite impressive. Naturally, established drugs such as Millennium Pharmaceuticals‘ (MLNM) Velcade, Genentech’s (DNA) Rituxan and Celgene’s (CELG) Revlimid got most of the attention.
In my opinion, the real star of the conference is MT-103, which is being co-developed by Micromet (MITI) and MedImmune, the biologics division of AstraZeneca (AZN). I won’t go too deep into describing the mechanism of action and the platform based on which MT-103 is built (I intend to do that in a review I hope to publish next week). However, the clinical data presented by Micromet is so impressive and so groundbreaking from several perspectives that it must not be ignored.
MT-103 is an antibody which is evaluated for the treatment of several blood cancers, including NHL (Non-Hodgkins Lymphoma). Similar to other antibodies evaluated for the treatment of cancer, MT-103 utilizes an antibody’s ability to specifically and selectively target cancer cells, however, it also represents a very different approach to targeting them. If we try to classify MT-103, it can be described by two primary factors: it is a (i) bi-specific antibody and a (ii) single-chain Fv antibody.
In the last decade, researchers have evaluated countless bi-specific antibodies as well as countless single-chain Fv antibodies. The vast majority of these candidates failed to show the slightest clinical activity, and consequently, the antibody industry decided to focus on more “conventional” antibody types. I must admit that when I first heard about MT-103, I was sure it would be a complete flop. I remember thinking to myself, “these guys take two niches of the antibody field that have been a clinical graveyard for years, combine them, and expect to get an effective drug – not going to happen”. Well, it happened big time. I will try to elaborate more about bi-specific antibodies and single-chain Fv antibodies later, but for now the most important thing to understand is that the combination of the two “underperforming” approaches is probably the reason for the astounding anti-cancer activity demonstrated by MT-103.
Even more astounding are the low doses of MT-103 that showed such an impressive clinical activity. This trial was a dose escalation trial, so it involved multiple doses with the intention of finding the highest possible dose that can be administered without causing unbearable side-effects. Although the research team has yet to identify the maximum tolerated dose, the 3 highest evaluated doses showed very promising results.
Among the 18 patients who received the 3 highest doses (0.015 to 0.06 mg/m2 per day), three had a complete response and four had a partial response, which leads to a response rate of 39%. This response rate is very impressive especially in light of the fact that most participants were heavily pre-treated patients in advanced stages of the disease. Moreover, all three patients who received the highest dose (0.06 mg/m2 per day) had a clinical response. These dose levels are substantially lower than those used for any other antibody-based therapy I have ever seen. A comparison to Rituxan, an approved antibody for the treatment of NHL might clarify that point.
Rituxan, which is the best selling antibody in history and is used to treat similar patient population, is generally dosed at 375 mg/m² per week. MT-103 was dosed every day, so in order to compare MT-103 doses to that of Rituxan, doses should be multiplied by 7. In addition, because MT-103 is much smaller than a typical antibody like Rituxan, the dose should be further multiplied by 3 to get an apples-to-apples comparison. Therefore, MT-103 was dosed at 0.315-1.26 mg/m2 per week, which are more than 300 fold lower than the typical dose of Rituxan. Historical clinical data shows that Rituxan leads to a response rate of 40-50% in relapsed patients, which makes the 39% response rate achieved by the extremely low doses of MT-103 very impressive.
Finally, a few words of caution. Investing in a tiny company such as Micromet, with an early-stage candidate such as MT-103 bears a very high risk, no matter how impressive preliminary clinical results are. The fact that MT-103 is a very unusual antibody platform certainly adds more uncertainty. Nevertheless, the impressive results among heavily pretreated lymphoma patients, the “homeopathic” doses, and the sheer size of MT-103’s addressable market lead us to the conclusion that Micromet represents a very attractive investment opportunity.
mfg ipollit
Micromet, Inc.: The Real Star of ASH 2007
by: Ohad Hammer posted on: December 14, 2007 | about stocks: MITI
A lot of clinical data was published at the American Society of Hematology [ASH] meeting, some of it quite impressive. Naturally, established drugs such as Millennium Pharmaceuticals‘ (MLNM) Velcade, Genentech’s (DNA) Rituxan and Celgene’s (CELG) Revlimid got most of the attention.
In my opinion, the real star of the conference is MT-103, which is being co-developed by Micromet (MITI) and MedImmune, the biologics division of AstraZeneca (AZN). I won’t go too deep into describing the mechanism of action and the platform based on which MT-103 is built (I intend to do that in a review I hope to publish next week). However, the clinical data presented by Micromet is so impressive and so groundbreaking from several perspectives that it must not be ignored.
MT-103 is an antibody which is evaluated for the treatment of several blood cancers, including NHL (Non-Hodgkins Lymphoma). Similar to other antibodies evaluated for the treatment of cancer, MT-103 utilizes an antibody’s ability to specifically and selectively target cancer cells, however, it also represents a very different approach to targeting them. If we try to classify MT-103, it can be described by two primary factors: it is a (i) bi-specific antibody and a (ii) single-chain Fv antibody.
In the last decade, researchers have evaluated countless bi-specific antibodies as well as countless single-chain Fv antibodies. The vast majority of these candidates failed to show the slightest clinical activity, and consequently, the antibody industry decided to focus on more “conventional” antibody types. I must admit that when I first heard about MT-103, I was sure it would be a complete flop. I remember thinking to myself, “these guys take two niches of the antibody field that have been a clinical graveyard for years, combine them, and expect to get an effective drug – not going to happen”. Well, it happened big time. I will try to elaborate more about bi-specific antibodies and single-chain Fv antibodies later, but for now the most important thing to understand is that the combination of the two “underperforming” approaches is probably the reason for the astounding anti-cancer activity demonstrated by MT-103.
Even more astounding are the low doses of MT-103 that showed such an impressive clinical activity. This trial was a dose escalation trial, so it involved multiple doses with the intention of finding the highest possible dose that can be administered without causing unbearable side-effects. Although the research team has yet to identify the maximum tolerated dose, the 3 highest evaluated doses showed very promising results.
Among the 18 patients who received the 3 highest doses (0.015 to 0.06 mg/m2 per day), three had a complete response and four had a partial response, which leads to a response rate of 39%. This response rate is very impressive especially in light of the fact that most participants were heavily pre-treated patients in advanced stages of the disease. Moreover, all three patients who received the highest dose (0.06 mg/m2 per day) had a clinical response. These dose levels are substantially lower than those used for any other antibody-based therapy I have ever seen. A comparison to Rituxan, an approved antibody for the treatment of NHL might clarify that point.
Rituxan, which is the best selling antibody in history and is used to treat similar patient population, is generally dosed at 375 mg/m² per week. MT-103 was dosed every day, so in order to compare MT-103 doses to that of Rituxan, doses should be multiplied by 7. In addition, because MT-103 is much smaller than a typical antibody like Rituxan, the dose should be further multiplied by 3 to get an apples-to-apples comparison. Therefore, MT-103 was dosed at 0.315-1.26 mg/m2 per week, which are more than 300 fold lower than the typical dose of Rituxan. Historical clinical data shows that Rituxan leads to a response rate of 40-50% in relapsed patients, which makes the 39% response rate achieved by the extremely low doses of MT-103 very impressive.
Finally, a few words of caution. Investing in a tiny company such as Micromet, with an early-stage candidate such as MT-103 bears a very high risk, no matter how impressive preliminary clinical results are. The fact that MT-103 is a very unusual antibody platform certainly adds more uncertainty. Nevertheless, the impressive results among heavily pretreated lymphoma patients, the “homeopathic” doses, and the sheer size of MT-103’s addressable market lead us to the conclusion that Micromet represents a very attractive investment opportunity.
mfg ipollit
Antwort auf Beitrag Nr.: 34.732.423 von ipollit am 15.08.08 01:14:17Micromet: Biting Cancer
by: Ohad Hammer posted on: February 06, 2008 | about stocks: IMGN / MITI / SGEN
Today it is clear that treating cancer with monoclonal antibodies is one of the greatest advancements in oncology. Just over a decade ago, the approval of Rituxan marked the birth of a multi-billion dollar market, as 8 additional antibodies have since joined Rituxan. The market is currently dominated by a specific flavor of antibodies termed “naked” antibodies, which represent a fraction of the large amount of different antibody flavors.
In contrast to naked antibodies, other flavors have yet to reach maturity, although some of these are making their way steadily to the center stage. All these approaches have one thing in common: They rely on antibodies’ exquisite ability to recognize and bind a target in a very specific manner. One of these approaches, represented by Immunogen (IMGN) and Seattle Genetics (SGEN), deals with Antibody-drug conjugates (ADCs), which are constructed by linking antibodies to a drug-payload. The antibody serves as a guiding system by guiding the drug to tumors, and releases it inside cancer cells.
In addition, there is a lot of activity in developing additional antibody-based therapies that involve linking other types of substances to antibodies. For example, one possibility for boosting an antibody’s potency is linking it to a radioactive molecule like in GlaxoSmithKline’s (GSK) Bexxar case.
In biotech, just like in other investment fields, it is important to recognize market trends, and identify emerging technologies and concepts. The problem with such cutting-edge technologies is that regardless of how promising they seem, there is always an unknown period of incubation, in which the technology migrates from basic research to the industry. If we take the whole antibody industry as an example, it took almost a quarter of a century from the scientific breakthrough that gave rise to monoclonal antibodies, to the approval of Rituxan. In the case of ADCs, several encouraging results may imply that the incubation period is finally over, although drug development is always characterized with a high level of uncertainty.
As someone who has been following the antibody market for quite some time now, I assumed that ADCs such as T-DM1 will represent the majority of clinical breakthroughs in the coming years. However, preliminary results from a small clinical trial that were published in ASH three months ago, showed that there is a unique platform which can generate highly potent antibodies, without even linking them to drugs or other effector molecules. In fact, this platform gave rise to one of the most potent antibodies ever tried on human beings - Micromet’s (MITI) MT103.
Micromet is a German-based company that developed a unique antibody-based platform termed BiTE (Bispecific T Cell Engagers). BiTE antibodies are unique in the sense that they combine two approaches that have been investigated for years without any meaningful success. I discussed the clinical activity of MT103 in brief here and intend to do so more thoroughly in the second part of this article. But first, let’s look at the two approaches which are represented by BiTE antibodies.
First and foremost, BiTE antibodies are bi-specific antibodies (bsAbs). Unlike standard antibodies, which have identical structure and function to naturally occurring antibodies, bsAbs are not something that can be usually seen in nature. A “normal” antibody is commonly represented as a “Y” shaped molecule (see picture). It has two identical “arms”, each can bind exactly the same target. Therefore, such antibodies are considered mono-specific. A bi-specific antibody can also be represented as a “Y” shaped molecule, but it has two different arms, each one capable of binding a different target. Hence, a bispecific antibody can simultaneously bind two different and unrelated targets. It doesn’t take too much imagination to understand that this type of antibodies theoretically opens new and exciting perspectives for antibody-based therapy.
In the context of cancer therapy, bispecific antibodies are usually designed to redirect immune cells to tumor cells, in a way that will lead to the attack of the tumor. One arm is directed against a target on a cancer cell while the other arm is directed against a target on the immune cell. In most cases, bispecific antibodies must not only bring the immune cell closer to cancer cells but also activate it to attack the tumor. In order to achieve both of these tasks, there is a need to identify specific targets on immune cells that can be activated by antibody binding, or find supplementary ways to achieve this activation.
Naturally, there is a large number of options when designing a bsAb. One variable is the target on the cancer cells, another variable is the type of immune cell to be recruited, while a third variable is which structural element (antigen) on that specific immune cell should be targeted. This is much more complicated than designing mono-specific antibodies, in which only the target on the cancer cells should be chosen.
Cancer cells are constantly created in our body but in the vast majority of cases, our immune system identifies and exterminates them. In some cases, however, a cancer cell manages to evade or suppress the immune response, thus enabling itself to multiply and spread throughout the body. BsAbs are similar to cancer vaccines such as Cell Genesys’ (CEGE) GVAX in their intent to promote an immune response against cancer. This approach holds that there is an immense potential in our immune system that can be unleashed upon cancer since our immune system commands a very potent and diverse arsenal of anti-cancer weapons. In both cases, the challenge is activating these weapons effectively and safely, with an emphasis on “safely”.
Manipulating our immune system is not something to be taken lightly, as it is compiled of many players and interactions, that are elegantly orchestrated. Because our immune system is so potent, it is also heavily regulated in order to keep things from getting out of hand. Auto-immune diseases, such as Rheumatoid arthritis and Multiple sclerosis, where the immune system attacks the body’s healthy tissues, are one example for what happens when this control is lost.
The phase I trial of TGN1412 in 2006 should serve as reminder for the explosive and dangerous potential immune modulating antibodies have. TGN1412 is an antibody that binds CD28, a receptor on T cells that can strongly activate them, upon antibody binding. This antibody was evaluated for safety in a phase I study that turned into one of the most notorious clinical trials in recent years. Apparently, TGN1412 led to a strong immune response that simply got out of control, sending immune cells rampaging through the patients’ bodies, destroying healthy tissues. All 6 patients who received this antibody were rushed to the ER in critical state with severe side effects that included severe pains, convulsing, and failure of the liver, heart and lungs.
Bispecific antibodies may prove to be safer than antibodies such as TGN1412 since they redirect the immune cells to respond specifically against cancer cells that express a specific target. Nevertheless, immune cells can communicate with each other to create a systemic response. This might be very positive when the systemic response is controlled and channeled against cancer, as it might overcome the immunosuppressant nature of tumors. Nevertheless, this might also lead to devastating results if the immune response is turned against normal tissues.
Bispecific antibodies are still a minor niche among the industry and as such, are ignored by most pharma companies, due to a history of technical difficulties and poor evidence of clinical activity. Will we ever see commercial drugs based bsAbs? Nobody knows, but Micromet is getting closer than any company has ever gotten to realizing the potential of bispecific antibodies.
The second structural feature of BiTE antibodies is their small size, about one third of normal antibodies like Rituxan and Herceptin. Small antibodies, generally referred to as scFv (Single Chain Variable Fragment) antibodies, are constructed by taking only the tips of the antibodies’ binding arms, which are the regions through which any antibody binds its target. The best analogy I could find in order to explain the difference between small antibodies and regular ones, is comparing antibodies to a human arm. An antibody can be described as two whole arms while a small antibody can be descried as just the palms.
Smaller antibodies have some flaws that hampered most efforts to develop drugs based on this antibody type. First, their small size leads to very fast clearance from the patient’s body, so it is very difficult to maintain a sufficient level of the antibody in the bloodstream over time. Second, most small antibodies have only one arm, which decreases their binding abilities. One significant advantage of small antibodies is their ability to penetrate tissues more efficiently. The tissue penetration factor is regarded as very important in the case of solid tumors, where tumors are typically a dense mass of cells that is very hard to penetrate, as opposed to blood cancers, where tumors are much more accessible. Moreover, in advanced cancers, tumors often metastasize by sending cancer cells that penetrate into the patient’s organs. These metastatic lesions are less approachable to conventional antibodies. Until now, the disadvantages in small antibodies outdid the advantages, but ironically, this feature has been crucial for the success of BiTE antibodies.
**********************************************
Micromet: Biting Cancer (Part II)
by: Ohad Hammer posted on: February 10, 2008 | about stocks: MITI
BiTE Antibodies
A BiTE (Bispecific T Cell Engager) antibody is a bi-specific antibody (bsAb) which directs T-cells to attack cancer cells, by simultaneously binding the two cells. Upon binding, a physical link is created between the two cells, which in turn triggers the T cell to attack the target cell. Every BiTE antibody has two binding arms, the first binds the CD3 receptor present on T-cells and the second binds a specific element on a cancer cell. The T-cell binding arm provides the activity while the cancer binding arm provides the specificity. By changing the cancer binding arm, the BiTE antibody can be adapted not only from one type of cancer to another, but also from one target to another in the same type of cancer. Therefore, BiTE represents a universal and modular platform for producing bsAbs for an unlimited number of targets.
As previously stated, bi-specific antibodies are aimed at recruiting immune cells against cancer. Therefore, one of the first decisions to be made concerns the type of immune cells to be recruited. The first attempts to develop bi-specific antibodies, mainly included recruiting T-cells, which are considered the most potent cells of the immune system. T cells play a critical role in the body’s efforts to eliminate malfunctioning cells such as cancer or virally infected cells, making them even more obvious candidates. T cells attack target cells by bombarding them with toxic proteins that eventually force the cell to commit programmed-cell death (apoptosis), and are considered very efficient in fighting cancer for several reasons. First, when properly activated, T cells act as serial killers, killing multiple cells one after the other. Second, upon activation, T cells start to multiply at the site of activation (which is hopefully next to the tumor), increasing the number of effector cells at an exponential rate. Third, T cells are constantly scanning tissues in order to find cancer cells. Many cancer cells manage to avoid being recognized by a T cell, even if it passes in proximity, however, a bsAb might provoke the T cell to recognize and attack even the most evasive of cancer cells.
Most of the efforts to trigger T cells against cancer cells utilized the CD3 receptor because of its involvement in the activation of T cells’ attacking mechanisms, but this concept remained invalidated for over twenty years, since no one was able to decipher exactly how T cells are triggered through this receptor. Today, an increasing body of scientific evidence, including one clinical trial, implies that Micromet has found the recipe for the activation of T cells via the CD3 receptor. So how did Micromet succeed where others have repeatedly failed?
It turned out that in order to achieve maximal stimulation of T-cells, they must be as close as possible to the cancer cell. While larger antibodies manage to bind a T cells and a cancer cell simultaneously, the distance between the two is not short enough for the T cell to “realize” it is standing in front of a cancer cell. Micromet’s BiTE antibodies are unique because they are much smaller and flexible and consequently bring T cells close enough to cancer cells to generate a powerful response.
The extremely high potency of BiTE antibodies was already observed in early pre-clinical trials, where BiTE antibodies demonstrated up to a 1000 fold increase in potency compared to prior bispecific antibodies that target CD3. BsAbs are commonly assessed by their E:T ratio. This number represents the ratio between the number of effector cells ( in our case, T cells) to the number of target cells (cancer cells) needed in order to show a substantial anti-cancer activity. Obviously, a more potent bsAb will have a lower E:T ratio, since it implies each immune cell kills more target cells at a given time. Most bsAbs have an E:T ratio of 40-100:1, whereas some which use CD3 have a ratio of 2:1. MT103, the first BiTE antibody to enter the clinic, is the only bispecific antibody I am familiar with which achieved a lower than 1 E:T ratio: A staggering 1:10 ratio. In other words, a potent anti-cancer activity is observed even if cancer cells outnumber T cells by a factor of 10!
MT103
MT103 targets CD19, a receptor which is expressed in many blood cancers including Non-Hodgkin’s Lymphoma [NHL] as well as several leukemias. Because CD19 is expressed in the majority of blood cancers, it is often described as “The next CD20″. Throughout the years there were several attempts to target CD19 with naked antibodies, but none of them bore fruit. Today, CD19 is receiving a great deal of attention in the pharma industry as several companies, including Sanofi-Aventis (SNY) and Genentech (DNA) are developing Antibody-drug conjugates based on anti-CD19 antibodies. Although none of the other companies who target CD19 reported clinical results, the CD19 market is going to be highly competitive.
In the first clinical trials for MT103, the antibody was given in a series of infusions similarly to the way standard antibodies are administered. These trials failed to show any significant clinical response and were associated with severe side effects that led to the early termination of the trials. Then, in 2004 Micromet started another phase I trial in NHL patients, in which MT103 was to be administered via continuous infusion. This is very unusual, as the typical dosing of antibodies for cancer is once a week. Conventional antibodies tend to stay in patients’ bloodstream for more than two weeks, and unlike MT103, are very safe at high doses. However, the company did not have much of a choice, and it hoped that by lengthening the infusion time, the side effects could be mitigated and the overall amount of administered MT103 could be increased. It is important to mention that patients do not need to be hospitalized throughout the course of treatment, but simply carry a small pump the size of a cell phone that constantly infuses them with the drug.
This strategy proved successful as side effects decreased substantially compared to the short infusion trials, and the company’s CEO boasts the fact that patients actually gained weight during the treatment, a great unofficial indicator for safety. More importantly, signs of clinical activity could be seen very early in the dose escalation process.
The primary objective in phase I trials is verifying the safety of the drug and not necessarily its clinical activity. As a result, these trials typically start from very low doses in order to make sure the drug is safe, so nobody really expects the smallest doses in phase I trials to have a therapeutic effect. Amazingly, the first patient that was enrolled to the trial achieved Stable disease [SD], followed by only 4 weeks of treatment. The study has enrolled over 30 patients in 6 different cohorts. Out of the nine Patients who received the next two doses (cohorts 2 & 3 ), eight had SD as well. The trend continued, as in cohorts 4-6 multiple partial and complete responses were observed.
It is still too early to reach conclusions as to the efficacy of MT103 in this trial since dose escalation is still ongoing, however, with 7 out of 20 patients in the three highest doses achieving clinical responses, it definitely looks like that MT103 has a great potential for the treatment of several subtypes of NHL. Moreover, all three patients who received the highest dose, had an objective response ( One had a CR and the other two had PRs). It won’t be reasonable to expect that this dose can actually achieve 100% response rate in larger populations, but this is indeed a great sign of a dose dependent activity.
While the response rate of this trial is very encouraging, the actual doses which led to the responses are nothing short of sensational. MT103 was effective at doses at which most antibody-based platforms are not even evaluated. The highest dose in the trial was 0.06 mg/m2 per day, which translates to 0.42 mg/m2 per week. Because MT103 is a small antibody, the actual number of antibody molecules per mg is three times higher, so in order to level the playfield, the dose of MT103 should be regarded as 1.26 mg/m2 per week. Rituxan, which is approved for the treatment of NHL, is administered at 375 mg/m2 per week, an almost 300 fold difference.
This comparison is brought just to give an idea for how groundbreaking the MT103 data really is. The two antibodies have never been compared and there are still a lot of data missing in order to reach any definitive conclusions. It also does not imply that MT103 is a direct competitor of Rituxan, since they target two different antigens and seem to have a different activity spectrum for different subtypes of NHL. Moreover, Rituxan was found to augment the activity of antibody platforms against CD19 in animal models, so combining the two antibodies is certainly an option. What we can safely say, though, is that MT103 showed a strong clinical effect in a small group of patients, 90% of whom had been previously treated with Rituxan. In addition, we can also say that Rituxan is very unlikely to have any effect when dosed anywhere near the MT103 levels.
NHL is a collection of several types of lymphomas, which may vary in their response to a given treatment. The phase I trial patient population was rather heterogeneous but several observations can be made about MT103’s activity among several subtypes of NHL.
The most evident effect was shown in patients with Mantle cell lymphoma [MCL]. This aggressive subtype of NHL represents about six percent of all NHL cases in the United States. The MT103 phase I trial included 15 MCL patients, but if we look again at the three highest doses, there were two complete responses and one partial response out of eight patients (response rate of 42%). Rituxan, for instance, has 27%-37% response rate in MCL as mono-therapy, but when added to chemotherapy regimens, response rate increases to 60%-90%. Despite these favorable response rates, most patients relapse after initial treatments, making MCL one of the most challenging lymphomas.
Another subgroup of patients who seemed to benefit from MT103 treatment is patients with Follicular lymphoma [FL], which is the second most frequent lymphoma worldwide. Of the eight FL patients in the three highest doses, one achieved complete response while the other two achieved partial response (response rate of 42%). FL has been proven to be very sensitive to Rituxan, with around 50% response rate in pre-treated patients. Patients whose disease relapse after the treatment with Rituxan are still responsive to the antibody, but with a lower response rate.
Micromet launched a Phase II trial in Acute Lymphoblastic Leukemia [ALL] patients in October 2007. ALL is an extremely aggressive disease characterized by high mortality rates. ALL patients are typically responsive to chemotherapy, but in many cases, a small number of cancer cells manage to survive in the bone marrow, leading to a very quick relapse after less than 6 months. In this phase II trial, MT103 is given to patients who achieve remission with standard treatments, but still have remnants of cancer in their bone marrow. The high activity of MT103 in targeting cancer cells inside the bone marrow, might prove useful in maintaining ALL patients in remission for longer periods.
The company stated it regards this trial as a proof-of-concept trial for MT103 in aggressive blood cancers. If proven effective, MT103 can find itself in a phase II trial for DLBCL, which is the most common aggressive lymphoma and therefore represents one of the biggest commercial opportunities for MT103. This trial is also used to validate the concept of using MT103 for decreasing chances of disease relapse after conventional therapy.
MedImmune (Now part of AstraZeneca) partnered with Micromet for the development of MT103 and is expected to launch two trials in the US. The first will enroll NHL patients, similarly to the original phase I trial reported at ASH. The second trial will focus on CLL, the most common leukemia in western countries. CLL cells are characterized by relatively low expression of CD20, Rituxan’s target, which can serve as an explanation why CLL is less sensitive to the antibody. In addition, A retrospect analysis among CLL patients showed that CD19 expression may be associated with higher risk of disease progression and death.
Despite MT103’s promising activity in blood cancers, we must not forget that most patients are treated with a combination of chemo/radio–therapy and monoclonal antibodies. There is no way to predict whether MT103 can be co-administered with standard therapies, despite the good safety profile. This combination might even prove to be somewhat counterproductive, as the potential proliferation of T cells can be offset by chemotherapy and radiation treatments.
Going forward, BiTE’s real potential lies in the treatment of solid tumors, which represent more than 90% of cases of cancer worldwide. Unfortunately, naked antibodies against solid tumors suffer from very poor success rates in the clinic, and even the approved ones have modest efficacy, when added to chemotherapy. Companies who develop antibodies for cancer prefer to focus on blood cancers, which are far more responsive and easy to treat. This will ultimately turn the market of antibodies for blood cancers into a very crowded and competitive one. The market of solid tumors will be characterized by higher entry barriers and lower success rates on the one hand, but also by a lack of competition and great demand even for slightly effective antibodies.
The main problem with conventional antibodies such as Herceptin and Erbitux
is that only a tiny fraction of the total injected antibody molecules actually reaches the tumors, and when they do, they have a very subtle effect, as each antibody affects one cancer cell, not necessarily killing it. In addition, conventional antibodies affect primarily the more external cells of the tumor, since they cannot penetrate the dense tumor mass.
These are some of the issues that should be confronted by next-generation antibody platforms, such as the BiTE platform. Micromet’s approach looks very appealing because it actually deals with several issues at once. First, BiTE antibodies have a better chance of killing a cancer cell upon binding, assuming they succeed in recruiting T cells. Second, BiTE antibodies are smaller so they might be able to penetrate solid tumors more easily. Third, the use of T cells as effector cells may lead to a very strong amplification, so it might take a small amount of antibodies to trigger a powerful immune response.
One of the most impressive cases in the MT103 trial was an MCL patient whose liver was infiltrated with cancer cells. This patient had a CR after four weeks of treatment, as MT103 managed to wipe out all detectable tumors in the liver, according to a biopsy. This is certainly not a case of metastatic cancer but it implies that MT103 can penetrate and generate a potent immune response also in less accessible sites, in addition to its activity in the bloodstream and bone marrow.
Nevertheless, we are dealing here with drug development, where for every reason why a certain drug should work, there are ten reasons why it shouldn’t. There is still no clinical data about BiTE antibodies for the treatment of solid tumors and historical success rates for solid cancers average around a mid single digit. Therefore, Micromet involves tremendous risks and uncertainties as well as great potential.
The company has more than five additional BiTE antibodies in pre-clinical studies, all of which target solid tumors. This strategy is a breath of fresh air in an industry where most companies prefer to focus on blood cancers. It is a great example for how the next generation of antibody platforms should be developed: Proof of concept in blood cancers and then a strong shift to solid tumors, where market opportunities are huge. It would be unrealistic to expect most of Micromet’s clinical programs, including MT103, to succeed, but Still, if Micromet gets it right with only one of its BiTE molecules for solid cancers, the financial reward will be enormous.
Micromet intends to advance its first BiTE antibody for the treatment of solid tumors to the clinic in the coming weeks. The antibody, MT110, targets EpCAM, which is one of the most known and well characterized tumor-associated antigens. A recent scientific paper published by Micromet, presents very impressive results in animal models regarding the efficacy and safety of MT110. It remains to be seen whether these results can be, at least, partially duplicated in clinical trials.
mfg ipollit
by: Ohad Hammer posted on: February 06, 2008 | about stocks: IMGN / MITI / SGEN
Today it is clear that treating cancer with monoclonal antibodies is one of the greatest advancements in oncology. Just over a decade ago, the approval of Rituxan marked the birth of a multi-billion dollar market, as 8 additional antibodies have since joined Rituxan. The market is currently dominated by a specific flavor of antibodies termed “naked” antibodies, which represent a fraction of the large amount of different antibody flavors.
In contrast to naked antibodies, other flavors have yet to reach maturity, although some of these are making their way steadily to the center stage. All these approaches have one thing in common: They rely on antibodies’ exquisite ability to recognize and bind a target in a very specific manner. One of these approaches, represented by Immunogen (IMGN) and Seattle Genetics (SGEN), deals with Antibody-drug conjugates (ADCs), which are constructed by linking antibodies to a drug-payload. The antibody serves as a guiding system by guiding the drug to tumors, and releases it inside cancer cells.
In addition, there is a lot of activity in developing additional antibody-based therapies that involve linking other types of substances to antibodies. For example, one possibility for boosting an antibody’s potency is linking it to a radioactive molecule like in GlaxoSmithKline’s (GSK) Bexxar case.
In biotech, just like in other investment fields, it is important to recognize market trends, and identify emerging technologies and concepts. The problem with such cutting-edge technologies is that regardless of how promising they seem, there is always an unknown period of incubation, in which the technology migrates from basic research to the industry. If we take the whole antibody industry as an example, it took almost a quarter of a century from the scientific breakthrough that gave rise to monoclonal antibodies, to the approval of Rituxan. In the case of ADCs, several encouraging results may imply that the incubation period is finally over, although drug development is always characterized with a high level of uncertainty.
As someone who has been following the antibody market for quite some time now, I assumed that ADCs such as T-DM1 will represent the majority of clinical breakthroughs in the coming years. However, preliminary results from a small clinical trial that were published in ASH three months ago, showed that there is a unique platform which can generate highly potent antibodies, without even linking them to drugs or other effector molecules. In fact, this platform gave rise to one of the most potent antibodies ever tried on human beings - Micromet’s (MITI) MT103.
Micromet is a German-based company that developed a unique antibody-based platform termed BiTE (Bispecific T Cell Engagers). BiTE antibodies are unique in the sense that they combine two approaches that have been investigated for years without any meaningful success. I discussed the clinical activity of MT103 in brief here and intend to do so more thoroughly in the second part of this article. But first, let’s look at the two approaches which are represented by BiTE antibodies.
First and foremost, BiTE antibodies are bi-specific antibodies (bsAbs). Unlike standard antibodies, which have identical structure and function to naturally occurring antibodies, bsAbs are not something that can be usually seen in nature. A “normal” antibody is commonly represented as a “Y” shaped molecule (see picture). It has two identical “arms”, each can bind exactly the same target. Therefore, such antibodies are considered mono-specific. A bi-specific antibody can also be represented as a “Y” shaped molecule, but it has two different arms, each one capable of binding a different target. Hence, a bispecific antibody can simultaneously bind two different and unrelated targets. It doesn’t take too much imagination to understand that this type of antibodies theoretically opens new and exciting perspectives for antibody-based therapy.
In the context of cancer therapy, bispecific antibodies are usually designed to redirect immune cells to tumor cells, in a way that will lead to the attack of the tumor. One arm is directed against a target on a cancer cell while the other arm is directed against a target on the immune cell. In most cases, bispecific antibodies must not only bring the immune cell closer to cancer cells but also activate it to attack the tumor. In order to achieve both of these tasks, there is a need to identify specific targets on immune cells that can be activated by antibody binding, or find supplementary ways to achieve this activation.
Naturally, there is a large number of options when designing a bsAb. One variable is the target on the cancer cells, another variable is the type of immune cell to be recruited, while a third variable is which structural element (antigen) on that specific immune cell should be targeted. This is much more complicated than designing mono-specific antibodies, in which only the target on the cancer cells should be chosen.
Cancer cells are constantly created in our body but in the vast majority of cases, our immune system identifies and exterminates them. In some cases, however, a cancer cell manages to evade or suppress the immune response, thus enabling itself to multiply and spread throughout the body. BsAbs are similar to cancer vaccines such as Cell Genesys’ (CEGE) GVAX in their intent to promote an immune response against cancer. This approach holds that there is an immense potential in our immune system that can be unleashed upon cancer since our immune system commands a very potent and diverse arsenal of anti-cancer weapons. In both cases, the challenge is activating these weapons effectively and safely, with an emphasis on “safely”.
Manipulating our immune system is not something to be taken lightly, as it is compiled of many players and interactions, that are elegantly orchestrated. Because our immune system is so potent, it is also heavily regulated in order to keep things from getting out of hand. Auto-immune diseases, such as Rheumatoid arthritis and Multiple sclerosis, where the immune system attacks the body’s healthy tissues, are one example for what happens when this control is lost.
The phase I trial of TGN1412 in 2006 should serve as reminder for the explosive and dangerous potential immune modulating antibodies have. TGN1412 is an antibody that binds CD28, a receptor on T cells that can strongly activate them, upon antibody binding. This antibody was evaluated for safety in a phase I study that turned into one of the most notorious clinical trials in recent years. Apparently, TGN1412 led to a strong immune response that simply got out of control, sending immune cells rampaging through the patients’ bodies, destroying healthy tissues. All 6 patients who received this antibody were rushed to the ER in critical state with severe side effects that included severe pains, convulsing, and failure of the liver, heart and lungs.
Bispecific antibodies may prove to be safer than antibodies such as TGN1412 since they redirect the immune cells to respond specifically against cancer cells that express a specific target. Nevertheless, immune cells can communicate with each other to create a systemic response. This might be very positive when the systemic response is controlled and channeled against cancer, as it might overcome the immunosuppressant nature of tumors. Nevertheless, this might also lead to devastating results if the immune response is turned against normal tissues.
Bispecific antibodies are still a minor niche among the industry and as such, are ignored by most pharma companies, due to a history of technical difficulties and poor evidence of clinical activity. Will we ever see commercial drugs based bsAbs? Nobody knows, but Micromet is getting closer than any company has ever gotten to realizing the potential of bispecific antibodies.
The second structural feature of BiTE antibodies is their small size, about one third of normal antibodies like Rituxan and Herceptin. Small antibodies, generally referred to as scFv (Single Chain Variable Fragment) antibodies, are constructed by taking only the tips of the antibodies’ binding arms, which are the regions through which any antibody binds its target. The best analogy I could find in order to explain the difference between small antibodies and regular ones, is comparing antibodies to a human arm. An antibody can be described as two whole arms while a small antibody can be descried as just the palms.
Smaller antibodies have some flaws that hampered most efforts to develop drugs based on this antibody type. First, their small size leads to very fast clearance from the patient’s body, so it is very difficult to maintain a sufficient level of the antibody in the bloodstream over time. Second, most small antibodies have only one arm, which decreases their binding abilities. One significant advantage of small antibodies is their ability to penetrate tissues more efficiently. The tissue penetration factor is regarded as very important in the case of solid tumors, where tumors are typically a dense mass of cells that is very hard to penetrate, as opposed to blood cancers, where tumors are much more accessible. Moreover, in advanced cancers, tumors often metastasize by sending cancer cells that penetrate into the patient’s organs. These metastatic lesions are less approachable to conventional antibodies. Until now, the disadvantages in small antibodies outdid the advantages, but ironically, this feature has been crucial for the success of BiTE antibodies.
**********************************************
Micromet: Biting Cancer (Part II)
by: Ohad Hammer posted on: February 10, 2008 | about stocks: MITI
BiTE Antibodies
A BiTE (Bispecific T Cell Engager) antibody is a bi-specific antibody (bsAb) which directs T-cells to attack cancer cells, by simultaneously binding the two cells. Upon binding, a physical link is created between the two cells, which in turn triggers the T cell to attack the target cell. Every BiTE antibody has two binding arms, the first binds the CD3 receptor present on T-cells and the second binds a specific element on a cancer cell. The T-cell binding arm provides the activity while the cancer binding arm provides the specificity. By changing the cancer binding arm, the BiTE antibody can be adapted not only from one type of cancer to another, but also from one target to another in the same type of cancer. Therefore, BiTE represents a universal and modular platform for producing bsAbs for an unlimited number of targets.
As previously stated, bi-specific antibodies are aimed at recruiting immune cells against cancer. Therefore, one of the first decisions to be made concerns the type of immune cells to be recruited. The first attempts to develop bi-specific antibodies, mainly included recruiting T-cells, which are considered the most potent cells of the immune system. T cells play a critical role in the body’s efforts to eliminate malfunctioning cells such as cancer or virally infected cells, making them even more obvious candidates. T cells attack target cells by bombarding them with toxic proteins that eventually force the cell to commit programmed-cell death (apoptosis), and are considered very efficient in fighting cancer for several reasons. First, when properly activated, T cells act as serial killers, killing multiple cells one after the other. Second, upon activation, T cells start to multiply at the site of activation (which is hopefully next to the tumor), increasing the number of effector cells at an exponential rate. Third, T cells are constantly scanning tissues in order to find cancer cells. Many cancer cells manage to avoid being recognized by a T cell, even if it passes in proximity, however, a bsAb might provoke the T cell to recognize and attack even the most evasive of cancer cells.
Most of the efforts to trigger T cells against cancer cells utilized the CD3 receptor because of its involvement in the activation of T cells’ attacking mechanisms, but this concept remained invalidated for over twenty years, since no one was able to decipher exactly how T cells are triggered through this receptor. Today, an increasing body of scientific evidence, including one clinical trial, implies that Micromet has found the recipe for the activation of T cells via the CD3 receptor. So how did Micromet succeed where others have repeatedly failed?
It turned out that in order to achieve maximal stimulation of T-cells, they must be as close as possible to the cancer cell. While larger antibodies manage to bind a T cells and a cancer cell simultaneously, the distance between the two is not short enough for the T cell to “realize” it is standing in front of a cancer cell. Micromet’s BiTE antibodies are unique because they are much smaller and flexible and consequently bring T cells close enough to cancer cells to generate a powerful response.
The extremely high potency of BiTE antibodies was already observed in early pre-clinical trials, where BiTE antibodies demonstrated up to a 1000 fold increase in potency compared to prior bispecific antibodies that target CD3. BsAbs are commonly assessed by their E:T ratio. This number represents the ratio between the number of effector cells ( in our case, T cells) to the number of target cells (cancer cells) needed in order to show a substantial anti-cancer activity. Obviously, a more potent bsAb will have a lower E:T ratio, since it implies each immune cell kills more target cells at a given time. Most bsAbs have an E:T ratio of 40-100:1, whereas some which use CD3 have a ratio of 2:1. MT103, the first BiTE antibody to enter the clinic, is the only bispecific antibody I am familiar with which achieved a lower than 1 E:T ratio: A staggering 1:10 ratio. In other words, a potent anti-cancer activity is observed even if cancer cells outnumber T cells by a factor of 10!
MT103
MT103 targets CD19, a receptor which is expressed in many blood cancers including Non-Hodgkin’s Lymphoma [NHL] as well as several leukemias. Because CD19 is expressed in the majority of blood cancers, it is often described as “The next CD20″. Throughout the years there were several attempts to target CD19 with naked antibodies, but none of them bore fruit. Today, CD19 is receiving a great deal of attention in the pharma industry as several companies, including Sanofi-Aventis (SNY) and Genentech (DNA) are developing Antibody-drug conjugates based on anti-CD19 antibodies. Although none of the other companies who target CD19 reported clinical results, the CD19 market is going to be highly competitive.
In the first clinical trials for MT103, the antibody was given in a series of infusions similarly to the way standard antibodies are administered. These trials failed to show any significant clinical response and were associated with severe side effects that led to the early termination of the trials. Then, in 2004 Micromet started another phase I trial in NHL patients, in which MT103 was to be administered via continuous infusion. This is very unusual, as the typical dosing of antibodies for cancer is once a week. Conventional antibodies tend to stay in patients’ bloodstream for more than two weeks, and unlike MT103, are very safe at high doses. However, the company did not have much of a choice, and it hoped that by lengthening the infusion time, the side effects could be mitigated and the overall amount of administered MT103 could be increased. It is important to mention that patients do not need to be hospitalized throughout the course of treatment, but simply carry a small pump the size of a cell phone that constantly infuses them with the drug.
This strategy proved successful as side effects decreased substantially compared to the short infusion trials, and the company’s CEO boasts the fact that patients actually gained weight during the treatment, a great unofficial indicator for safety. More importantly, signs of clinical activity could be seen very early in the dose escalation process.
The primary objective in phase I trials is verifying the safety of the drug and not necessarily its clinical activity. As a result, these trials typically start from very low doses in order to make sure the drug is safe, so nobody really expects the smallest doses in phase I trials to have a therapeutic effect. Amazingly, the first patient that was enrolled to the trial achieved Stable disease [SD], followed by only 4 weeks of treatment. The study has enrolled over 30 patients in 6 different cohorts. Out of the nine Patients who received the next two doses (cohorts 2 & 3 ), eight had SD as well. The trend continued, as in cohorts 4-6 multiple partial and complete responses were observed.
It is still too early to reach conclusions as to the efficacy of MT103 in this trial since dose escalation is still ongoing, however, with 7 out of 20 patients in the three highest doses achieving clinical responses, it definitely looks like that MT103 has a great potential for the treatment of several subtypes of NHL. Moreover, all three patients who received the highest dose, had an objective response ( One had a CR and the other two had PRs). It won’t be reasonable to expect that this dose can actually achieve 100% response rate in larger populations, but this is indeed a great sign of a dose dependent activity.
While the response rate of this trial is very encouraging, the actual doses which led to the responses are nothing short of sensational. MT103 was effective at doses at which most antibody-based platforms are not even evaluated. The highest dose in the trial was 0.06 mg/m2 per day, which translates to 0.42 mg/m2 per week. Because MT103 is a small antibody, the actual number of antibody molecules per mg is three times higher, so in order to level the playfield, the dose of MT103 should be regarded as 1.26 mg/m2 per week. Rituxan, which is approved for the treatment of NHL, is administered at 375 mg/m2 per week, an almost 300 fold difference.
This comparison is brought just to give an idea for how groundbreaking the MT103 data really is. The two antibodies have never been compared and there are still a lot of data missing in order to reach any definitive conclusions. It also does not imply that MT103 is a direct competitor of Rituxan, since they target two different antigens and seem to have a different activity spectrum for different subtypes of NHL. Moreover, Rituxan was found to augment the activity of antibody platforms against CD19 in animal models, so combining the two antibodies is certainly an option. What we can safely say, though, is that MT103 showed a strong clinical effect in a small group of patients, 90% of whom had been previously treated with Rituxan. In addition, we can also say that Rituxan is very unlikely to have any effect when dosed anywhere near the MT103 levels.
NHL is a collection of several types of lymphomas, which may vary in their response to a given treatment. The phase I trial patient population was rather heterogeneous but several observations can be made about MT103’s activity among several subtypes of NHL.
The most evident effect was shown in patients with Mantle cell lymphoma [MCL]. This aggressive subtype of NHL represents about six percent of all NHL cases in the United States. The MT103 phase I trial included 15 MCL patients, but if we look again at the three highest doses, there were two complete responses and one partial response out of eight patients (response rate of 42%). Rituxan, for instance, has 27%-37% response rate in MCL as mono-therapy, but when added to chemotherapy regimens, response rate increases to 60%-90%. Despite these favorable response rates, most patients relapse after initial treatments, making MCL one of the most challenging lymphomas.
Another subgroup of patients who seemed to benefit from MT103 treatment is patients with Follicular lymphoma [FL], which is the second most frequent lymphoma worldwide. Of the eight FL patients in the three highest doses, one achieved complete response while the other two achieved partial response (response rate of 42%). FL has been proven to be very sensitive to Rituxan, with around 50% response rate in pre-treated patients. Patients whose disease relapse after the treatment with Rituxan are still responsive to the antibody, but with a lower response rate.
Micromet launched a Phase II trial in Acute Lymphoblastic Leukemia [ALL] patients in October 2007. ALL is an extremely aggressive disease characterized by high mortality rates. ALL patients are typically responsive to chemotherapy, but in many cases, a small number of cancer cells manage to survive in the bone marrow, leading to a very quick relapse after less than 6 months. In this phase II trial, MT103 is given to patients who achieve remission with standard treatments, but still have remnants of cancer in their bone marrow. The high activity of MT103 in targeting cancer cells inside the bone marrow, might prove useful in maintaining ALL patients in remission for longer periods.
The company stated it regards this trial as a proof-of-concept trial for MT103 in aggressive blood cancers. If proven effective, MT103 can find itself in a phase II trial for DLBCL, which is the most common aggressive lymphoma and therefore represents one of the biggest commercial opportunities for MT103. This trial is also used to validate the concept of using MT103 for decreasing chances of disease relapse after conventional therapy.
MedImmune (Now part of AstraZeneca) partnered with Micromet for the development of MT103 and is expected to launch two trials in the US. The first will enroll NHL patients, similarly to the original phase I trial reported at ASH. The second trial will focus on CLL, the most common leukemia in western countries. CLL cells are characterized by relatively low expression of CD20, Rituxan’s target, which can serve as an explanation why CLL is less sensitive to the antibody. In addition, A retrospect analysis among CLL patients showed that CD19 expression may be associated with higher risk of disease progression and death.
Despite MT103’s promising activity in blood cancers, we must not forget that most patients are treated with a combination of chemo/radio–therapy and monoclonal antibodies. There is no way to predict whether MT103 can be co-administered with standard therapies, despite the good safety profile. This combination might even prove to be somewhat counterproductive, as the potential proliferation of T cells can be offset by chemotherapy and radiation treatments.
Going forward, BiTE’s real potential lies in the treatment of solid tumors, which represent more than 90% of cases of cancer worldwide. Unfortunately, naked antibodies against solid tumors suffer from very poor success rates in the clinic, and even the approved ones have modest efficacy, when added to chemotherapy. Companies who develop antibodies for cancer prefer to focus on blood cancers, which are far more responsive and easy to treat. This will ultimately turn the market of antibodies for blood cancers into a very crowded and competitive one. The market of solid tumors will be characterized by higher entry barriers and lower success rates on the one hand, but also by a lack of competition and great demand even for slightly effective antibodies.
The main problem with conventional antibodies such as Herceptin and Erbitux
is that only a tiny fraction of the total injected antibody molecules actually reaches the tumors, and when they do, they have a very subtle effect, as each antibody affects one cancer cell, not necessarily killing it. In addition, conventional antibodies affect primarily the more external cells of the tumor, since they cannot penetrate the dense tumor mass.
These are some of the issues that should be confronted by next-generation antibody platforms, such as the BiTE platform. Micromet’s approach looks very appealing because it actually deals with several issues at once. First, BiTE antibodies have a better chance of killing a cancer cell upon binding, assuming they succeed in recruiting T cells. Second, BiTE antibodies are smaller so they might be able to penetrate solid tumors more easily. Third, the use of T cells as effector cells may lead to a very strong amplification, so it might take a small amount of antibodies to trigger a powerful immune response.
One of the most impressive cases in the MT103 trial was an MCL patient whose liver was infiltrated with cancer cells. This patient had a CR after four weeks of treatment, as MT103 managed to wipe out all detectable tumors in the liver, according to a biopsy. This is certainly not a case of metastatic cancer but it implies that MT103 can penetrate and generate a potent immune response also in less accessible sites, in addition to its activity in the bloodstream and bone marrow.
Nevertheless, we are dealing here with drug development, where for every reason why a certain drug should work, there are ten reasons why it shouldn’t. There is still no clinical data about BiTE antibodies for the treatment of solid tumors and historical success rates for solid cancers average around a mid single digit. Therefore, Micromet involves tremendous risks and uncertainties as well as great potential.
The company has more than five additional BiTE antibodies in pre-clinical studies, all of which target solid tumors. This strategy is a breath of fresh air in an industry where most companies prefer to focus on blood cancers. It is a great example for how the next generation of antibody platforms should be developed: Proof of concept in blood cancers and then a strong shift to solid tumors, where market opportunities are huge. It would be unrealistic to expect most of Micromet’s clinical programs, including MT103, to succeed, but Still, if Micromet gets it right with only one of its BiTE molecules for solid cancers, the financial reward will be enormous.
Micromet intends to advance its first BiTE antibody for the treatment of solid tumors to the clinic in the coming weeks. The antibody, MT110, targets EpCAM, which is one of the most known and well characterized tumor-associated antigens. A recent scientific paper published by Micromet, presents very impressive results in animal models regarding the efficacy and safety of MT110. It remains to be seen whether these results can be, at least, partially duplicated in clinical trials.
mfg ipollit
im positiven Biotech-Sentiment heute Onyx besonders stark... Gap geschlossen

Players grab Onyx Pharma calls on tech breakout
Thu Aug 14, 2008 7:12pm EDT
By Doris Frankel
CHICAGO, Aug 14 (Reuters) - Many players snapped up call options in Onyx Pharmaceuticals Inc (ONXX.O: Quote, Profile, Research, Stock Buzz) on Thursday in hopes that the strength in the drugmaker's stock would continue after it broke through a key technical level.
Onyx shares jumped 9.5 percent to close at $44.79 on the Nasdaq, its highest level since mid-February. Its option volume was also brisk, running six times the normal level.
Onyx pierced a key technical level when it moved through its July highs near $42.50. It also filled a large gap on the chart that was left when the stock gapped lower on Feb. 15.
Those two developments might be viewed as a positive because it suggests that the bullish trend is intact and there might be more room to the upside, said Frederic Ruffy, options analyst at website WhatsTrading.com.
"There is a lot of optimism in the biotech sector," said Joseph Cusick, senior market analyst at online brokerage optionsXpress Holdings.
"It could be mergers and acquisitions in the space. It could be new drugs getting further along in the approval process in the pipeline," Cusick said.
In Onyx, for example, Wellington Management this week disclosed a stake in the company, he said.
Wellington said in a regulatory filing on Monday that it holds a 10.7 percent stake in Onyx's stock. The fund did not reveal when it acquired the stake or the purpose behind the transactions.
In the options market, 25,000 Onyx calls traded compared to 16,000 puts, according to option analytics firm Trade Alert. One of the busiest options were the September $50 call strikes, allowing players to buy Onyx shares at $50 apiece.
A call allows an investor to buy the company's shares at a given price and time while a put conveys the right to sell the security.
The implied volatility, a key driver of an options price, on all of Onyx options also spiked to around mid-80 percent, Ruffy said, a move that suggests more stock movement.
"The call buying in Onyx is probably benefiting from the recent M&A interest in the sector," said Medora Lee, editor/researcher at Web information site optionmonster.com.
Biotechnology company ImClone Systems Inc (IMCL.O: Quote, Profile, Research, Stock Buzz), which rebuffed a $60-per-share takeover offer from Bristol-Myers Squibb Co (BMY.N: Quote, Profile, Research, Stock Buzz), on Thursday hired JP Morgan Chase & Co (JPM.N: Quote, Profile, Research, Stock Buzz) as its financial adviser.
Onyx has also been mentioned as a potential takeover play many times, with German partner Bayer AG (BAYG.DE: Quote, Profile, Research, Stock Buzz) cited as a possible suitor, Ruffy said. (Reporting by Doris Frankel, editing by Mark Porter)
mfg ipollit
Players grab Onyx Pharma calls on tech breakout
Thu Aug 14, 2008 7:12pm EDT
By Doris Frankel
CHICAGO, Aug 14 (Reuters) - Many players snapped up call options in Onyx Pharmaceuticals Inc (ONXX.O: Quote, Profile, Research, Stock Buzz) on Thursday in hopes that the strength in the drugmaker's stock would continue after it broke through a key technical level.
Onyx shares jumped 9.5 percent to close at $44.79 on the Nasdaq, its highest level since mid-February. Its option volume was also brisk, running six times the normal level.
Onyx pierced a key technical level when it moved through its July highs near $42.50. It also filled a large gap on the chart that was left when the stock gapped lower on Feb. 15.
Those two developments might be viewed as a positive because it suggests that the bullish trend is intact and there might be more room to the upside, said Frederic Ruffy, options analyst at website WhatsTrading.com.
"There is a lot of optimism in the biotech sector," said Joseph Cusick, senior market analyst at online brokerage optionsXpress Holdings.
"It could be mergers and acquisitions in the space. It could be new drugs getting further along in the approval process in the pipeline," Cusick said.
In Onyx, for example, Wellington Management this week disclosed a stake in the company, he said.
Wellington said in a regulatory filing on Monday that it holds a 10.7 percent stake in Onyx's stock. The fund did not reveal when it acquired the stake or the purpose behind the transactions.
In the options market, 25,000 Onyx calls traded compared to 16,000 puts, according to option analytics firm Trade Alert. One of the busiest options were the September $50 call strikes, allowing players to buy Onyx shares at $50 apiece.
A call allows an investor to buy the company's shares at a given price and time while a put conveys the right to sell the security.
The implied volatility, a key driver of an options price, on all of Onyx options also spiked to around mid-80 percent, Ruffy said, a move that suggests more stock movement.
"The call buying in Onyx is probably benefiting from the recent M&A interest in the sector," said Medora Lee, editor/researcher at Web information site optionmonster.com.
Biotechnology company ImClone Systems Inc (IMCL.O: Quote, Profile, Research, Stock Buzz), which rebuffed a $60-per-share takeover offer from Bristol-Myers Squibb Co (BMY.N: Quote, Profile, Research, Stock Buzz), on Thursday hired JP Morgan Chase & Co (JPM.N: Quote, Profile, Research, Stock Buzz) as its financial adviser.
Onyx has also been mentioned as a potential takeover play many times, with German partner Bayer AG (BAYG.DE: Quote, Profile, Research, Stock Buzz) cited as a possible suitor, Ruffy said. (Reporting by Doris Frankel, editing by Mark Porter)
mfg ipollit
Antwort auf Beitrag Nr.: 34.731.424 von ipollit am 14.08.08 21:45:15nochmal Micromet...
Drug that uses the body's cells to blast cancer
By Fiona Macrae
Last updated at 11:58 PM on 14th August 2008
Cancer patients have seen their tumours blasted into submission by a new drug which harnesses the power of their own immune cells.
The 'serial killer' treatment completely eliminated some tumours and shrunk others resistant to existing therapies.
Further successful trials could lead to blinatumomab being on the market in less than five years.
The tests were carried out on patients with non-Hodgkin's lymphoma, but it is hoped the methods can be adapted to tackle other cancers.
The results have been described as an 'exciting' development in the use of immunotherapy, the process of using the body's own immune system to fight disease.
The drug treats non-Hodgkin's lymphoma - a cancer of the immune system - by glueing cancer-killing white blood cells called cytotoxic T cells to the tumour.
Once there, they release a poison that destroys the cancer.
Researcher Dr Patrick Baeuerle said: 'These cells circulate in our body stuffed with ammunition - little vesicles filled with toxins.
'It is just a matter of attaching them to the tumour cells and making them fire.
'The drug tickles the T cell in a very special way so that it fires and won't stop firing until the tumour cells have gone. It turns them into serial killers.'
Professor Peter Johnson, Cancer Research UK's chief clinician, said: 'These exciting preliminary results come from using them to harness the body's own immune response in a new way.
'Although the side effects need to be monitored carefully, we hope that this type of treatment will prove to be active in larger trials in the future.'
Dr Baeuerle gave the drug to 38 people who had stopped responding to conventional treatments for non-Hodgkin's lymphoma, a disease that affects 10,000 Britons a year and kills almost 4,500.
In four cases, the tumours were eliminated, with one patient still cancer-free more than a year later.
In another seven cases, the tumours shrunk considerably, the journal Science reports.
Dr Baeuerle, of Micromet, the German biotech company developing the drug, said: 'All the patients we treated had run out of therapy options, they were considered incurable.
'This study could potentially offer something new to patients and give them a longer time to live or other improvement.'
The drug is now being tested on patients with an aggressive form of blood cancer and similar compounds are being developed to tackle other cancers.
Sufferers of non-Hodgkin's lymphoma-include Michael Aspel, who was diagnosed with a slow-growing form of the disease in 2003.
For reasons that are not understood, the number of cases has been slowly but steadily rising for the last 50 years.
If the trend continues, it could be as common as breast or lung cancer by 2025.
Dr Cassian Yee, of the Fred Hutchinson Cancer Research Center, Seattle, who has had significant results using an alternative method of treating patients with white blood cells grown in the lab, said: 'This a significant study.
'It remains to be seen if most of the responses are long lasting. Certainly the results are very promising.'
mfg ipollit
Drug that uses the body's cells to blast cancer
By Fiona Macrae
Last updated at 11:58 PM on 14th August 2008
Cancer patients have seen their tumours blasted into submission by a new drug which harnesses the power of their own immune cells.
The 'serial killer' treatment completely eliminated some tumours and shrunk others resistant to existing therapies.
Further successful trials could lead to blinatumomab being on the market in less than five years.
The tests were carried out on patients with non-Hodgkin's lymphoma, but it is hoped the methods can be adapted to tackle other cancers.
The results have been described as an 'exciting' development in the use of immunotherapy, the process of using the body's own immune system to fight disease.
The drug treats non-Hodgkin's lymphoma - a cancer of the immune system - by glueing cancer-killing white blood cells called cytotoxic T cells to the tumour.
Once there, they release a poison that destroys the cancer.
Researcher Dr Patrick Baeuerle said: 'These cells circulate in our body stuffed with ammunition - little vesicles filled with toxins.
'It is just a matter of attaching them to the tumour cells and making them fire.
'The drug tickles the T cell in a very special way so that it fires and won't stop firing until the tumour cells have gone. It turns them into serial killers.'
Professor Peter Johnson, Cancer Research UK's chief clinician, said: 'These exciting preliminary results come from using them to harness the body's own immune response in a new way.
'Although the side effects need to be monitored carefully, we hope that this type of treatment will prove to be active in larger trials in the future.'
Dr Baeuerle gave the drug to 38 people who had stopped responding to conventional treatments for non-Hodgkin's lymphoma, a disease that affects 10,000 Britons a year and kills almost 4,500.
In four cases, the tumours were eliminated, with one patient still cancer-free more than a year later.
In another seven cases, the tumours shrunk considerably, the journal Science reports.
Dr Baeuerle, of Micromet, the German biotech company developing the drug, said: 'All the patients we treated had run out of therapy options, they were considered incurable.
'This study could potentially offer something new to patients and give them a longer time to live or other improvement.'
The drug is now being tested on patients with an aggressive form of blood cancer and similar compounds are being developed to tackle other cancers.
Sufferers of non-Hodgkin's lymphoma-include Michael Aspel, who was diagnosed with a slow-growing form of the disease in 2003.
For reasons that are not understood, the number of cases has been slowly but steadily rising for the last 50 years.
If the trend continues, it could be as common as breast or lung cancer by 2025.
Dr Cassian Yee, of the Fred Hutchinson Cancer Research Center, Seattle, who has had significant results using an alternative method of treating patients with white blood cells grown in the lab, said: 'This a significant study.
'It remains to be seen if most of the responses are long lasting. Certainly the results are very promising.'
mfg ipollit
Antwort auf Beitrag Nr.: 34.730.902 von ipollit am 14.08.08 20:37:13Biotechs als die aktuell einzigen Momentum-Player?
Biotech Stocks To Rise On Momentum
By ROB CURRAN
Of DOW JONES NEWSWIRES
NEW YORK -- Forget oil and commodities, biotechnology stocks are the new targets of momentum players, and that should drive up the stocks for weeks and months.
Market participants say stock and commodity markets are increasingly swayed by a rambling posse of short-term investors dubbed "momentum," or "momo," traders. Once a sector or commodity price starts to beat the broad market, it quickly draws hedge funds and proprietary desks desperate for something that works, without wasting time on fundamental analysis.
There's so much of this momentum buying that it perpetuates and accentuates that seeming outperformance.
On a fundamental basis, the biotechnology sector has little in common with the energy and commodities sectors. Lining up a chart of the Biotech HOLDRs Trust (BBH) with the Energy Select Sector SPDR (XLE), a basket of energy stocks, however, suggests that the same pool of capital that drove the XLE to record peaks in May has recently rushed into the biotech stocks that comprise the BBH so that it's approaching records of its own. Since July 1, the XLE has fallen 18%, while the BBH has risen 20%.
At around $203, the BBH is up 26% for 2008 so far and at its highest level since 2000, even as the Standard & Poor's 500 is down 13% and the XLE is off 8.1%. With that kind of "relative strength," the BBH should break through its all-time highs soon, according to the charts.
Sure, there are fundamental reasons for the biotech rally: On July 21, Roche Holding (RHHBY) offered to buy the 44% of Genentech (DNA) it doesn't already own, giving Genentech shares a lift of more than 25% since then. In a similar move, Bristol-Myers Squibb (BMY) offered to buy out the remainder of ImClone Systems (IMCL) for an even richer premium.
Yet merger and acquisition speculation can't fully account for gains in stocks like Celgene (CELG), at $75 and up 63% so far this year after trading as low as $60 on June 24. Some of the fundamental reports from the sector are worrying: promising Alzheimer's disease and multiple-sclerosis drugs from Irish player Elan (ELN) have run into safety concerns.
"It really has nothing to do with the underlying facts and figures," said Carter Worth, chief market technician at Oppenheimer. "It's the gambling nature of certain capital, coupled with the frustration of nothing else working."
The volatility of biotech stocks attracts the same "gambling" style of capital as certain commodities and stocks, Worth said. He compared the faddish nature of speculation in recent months -- remember the fertilizer boom? -- with that of 19th century America.
"One of the biggest speculative endeavors was the hunt for whale oil," Worth said. Apart from the rise of crude oil, "one thing that killed it off was the gold rush. The same kind of guy that goes out to sea for two year was lured into the gold rush."
Right now, the biotech sector is the only speculative one showing signs of life, Worth said. That momentum is likely to keep biotech running for weeks, or months, he said.
"If we were to characterize the movement of capital into biotech of late as a fad, well, fads have duration," the technician said.
On a longer-term basis, however, market participants warn that playing with biotech is playing with fire.
"This rally is overdue and should continue," said Marc Pado, market strategist at Cantor Fitzgerald. The industry depends on funding from venture capital, he said. "Those that don't hit on drugs go "bye-bye," and those that do get a bid."
mfg ipollit
Biotech Stocks To Rise On Momentum
By ROB CURRAN
Of DOW JONES NEWSWIRES
NEW YORK -- Forget oil and commodities, biotechnology stocks are the new targets of momentum players, and that should drive up the stocks for weeks and months.
Market participants say stock and commodity markets are increasingly swayed by a rambling posse of short-term investors dubbed "momentum," or "momo," traders. Once a sector or commodity price starts to beat the broad market, it quickly draws hedge funds and proprietary desks desperate for something that works, without wasting time on fundamental analysis.
There's so much of this momentum buying that it perpetuates and accentuates that seeming outperformance.
On a fundamental basis, the biotechnology sector has little in common with the energy and commodities sectors. Lining up a chart of the Biotech HOLDRs Trust (BBH) with the Energy Select Sector SPDR (XLE), a basket of energy stocks, however, suggests that the same pool of capital that drove the XLE to record peaks in May has recently rushed into the biotech stocks that comprise the BBH so that it's approaching records of its own. Since July 1, the XLE has fallen 18%, while the BBH has risen 20%.
At around $203, the BBH is up 26% for 2008 so far and at its highest level since 2000, even as the Standard & Poor's 500 is down 13% and the XLE is off 8.1%. With that kind of "relative strength," the BBH should break through its all-time highs soon, according to the charts.
Sure, there are fundamental reasons for the biotech rally: On July 21, Roche Holding (RHHBY) offered to buy the 44% of Genentech (DNA) it doesn't already own, giving Genentech shares a lift of more than 25% since then. In a similar move, Bristol-Myers Squibb (BMY) offered to buy out the remainder of ImClone Systems (IMCL) for an even richer premium.
Yet merger and acquisition speculation can't fully account for gains in stocks like Celgene (CELG), at $75 and up 63% so far this year after trading as low as $60 on June 24. Some of the fundamental reports from the sector are worrying: promising Alzheimer's disease and multiple-sclerosis drugs from Irish player Elan (ELN) have run into safety concerns.
"It really has nothing to do with the underlying facts and figures," said Carter Worth, chief market technician at Oppenheimer. "It's the gambling nature of certain capital, coupled with the frustration of nothing else working."
The volatility of biotech stocks attracts the same "gambling" style of capital as certain commodities and stocks, Worth said. He compared the faddish nature of speculation in recent months -- remember the fertilizer boom? -- with that of 19th century America.
"One of the biggest speculative endeavors was the hunt for whale oil," Worth said. Apart from the rise of crude oil, "one thing that killed it off was the gold rush. The same kind of guy that goes out to sea for two year was lured into the gold rush."
Right now, the biotech sector is the only speculative one showing signs of life, Worth said. That momentum is likely to keep biotech running for weeks, or months, he said.
"If we were to characterize the movement of capital into biotech of late as a fad, well, fads have duration," the technician said.
On a longer-term basis, however, market participants warn that playing with biotech is playing with fire.
"This rally is overdue and should continue," said Marc Pado, market strategist at Cantor Fitzgerald. The industry depends on funding from venture capital, he said. "Those that don't hit on drugs go "bye-bye," and those that do get a bid."
mfg ipollit
5 Potential Buyout Targets in Biotech - Barron's
by: SA Editor Rachael Granby posted on: August 17, 2008
Bristol-Myers Squibb (BMY) made a $4.5B bid for the shares of ImClone (IMCL) it doesn't already own. Roche Holdings (RHHBY.PK) made a $44B offer for the shares of Genetech (DNA) it doesn't own. Both targets want the bid prices raised. Barron's Lawrence Strauss says these are just the two latest examples of an increasingly common biotech strategy to use mergers, buyouts, and takeovers as a way to fatten product pipeline (and profit) by acquiring promising and pre-tested biotech drugs.
Big Pharma is feeling the need to find new products with blockbuster potential as several important drugs approach the expiration of their patent protection. This, of course, will open the market to cheaper, generic versions of the drug, cutting into Big Pharma profit. Leading the pack will be Pfizer's (PFE) cholesterol drug, Lipitor, which generated more than $12B in global sales last year. Other companies with expiring blockbuster patents include Wyeth (WYE), Merck (MRK), and Eli Lilly (LLY). Jay Markowitz, a T. Rowe Price health-care analyst, notes "a number of companies are facing a significant patent cliff, where billions in revenues are going to disappear."
Many large pharmaceuticals have lots of cash on their balance sheets, making acquisitions an affordable option. The weaker dollar has also made U.S. companies look more attractive to biotech and pharmaceutical firms abroad. Another major factor is the difficult process of receiving FDA approval. This lengthy and grueling process provides an additional incentive to buy companies that have already received FDA approval on their drugs, ensuring smooth pipeline production going forward.
Potential buyout targets to keep an eye on: Amylin Pharmaceuticals (AMLN), United Therapeutics (UTHR), Alexion Pharmaceuticals (ALXN), Onyx Pharmaceuticals (ONXX), Vertex Pharmaceuticals (VRTX).
*********
Thapar and other analysts point to Onyx Pharmaceuticals (ONXX) as a potential target of Bayer. Onyx has a joint venture with the German pharmaceutical giant for Nexavar, which is used to treat kidney and liver cancers. "At some point, you could see Bayer wanting to control 100% of the assets," says Sustersic, who thinks that Bayer might pay as much as $65 per share. Nexavar is believed to have more upside, especially as it secures approval in other markets (it was recently given the green light by China as a liver-cancer treatment), and even more if it can be used for other cancers, including those of the breast, lungs and skin.
Not all of the focus is on products already in the market, however. Vertex Pharmaceuticals (VRTX), another company that some investors view as an eventual takeover candidate, is developing Telaprevir, a substance that promises to reduce the time needed to treat hepatitis-C, a potentially fatal liver ailment. Telaprevir is currently in stage III testing, the last phase of the clinical- trial process. Vertex has worked with Johnson & Johnson (JNJ) in developing that drug.
Another company that could spark an acquirer's interest is Amylin Pharmaceuticals (AMLN). It's working on a Type 2 diabetes drug that could be injected once a week instead of daily. Amylin has several development partners, including Eli Lilly, on the drug. It already markets two diabetes drugs, Symlin and Byetta. But some analysts say the product under development could boast major advantages. "A once-weekly drug that lowers glucose substantially, induces weight loss, isn't associated with hypoglycemia, and lacks a cardiovascular safety signal has multibillion-dollar potential," says Markowitz.
Anyone scanning the biotech ranks for potential acquisition targets shouldn't overlook United Therapeutics (UTHR). It has one characteristic that lots of other small biotechs would envy: It's in the black. One of its most promising products is Remodulin, which is used to treat hypertension in the blood vessels of the lungs. Although there are other PAH drugs, Turner Investment Partners' Sustersic says that United Therapeutics is "one of the clear leaders, and they potentially could have a big blockbuster."
American Century's Thapar is similarly impressed by Alexion Pharmaceuticals (ALXN), which earned six cents a share in its most recent quarter, versus a loss of 75 cents a share a year earlier. Its drugs include Soliris, which treats a rare disorder called paroxysmal nocturnal hemoglobinuria, which destroys red blood cells. At $389,000 a year per patient, Soliris is hugely expensive -- but it targets a clearly defined patient base. The drug had net sales of nearly $60 million in the second quarter, up from $45.5 million in the first quarter and $9.8 million a year earlier. Regulators have approved its use in the U.S. and Europe, and it's expected to be introduced in Japan toward the end of next year. Alexion retains distribution rights in the U.S. and overseas, says Sustersic, who maintains that Soliris sales could reach at least $500 million a year.
mfg ipollit
by: SA Editor Rachael Granby posted on: August 17, 2008
Bristol-Myers Squibb (BMY) made a $4.5B bid for the shares of ImClone (IMCL) it doesn't already own. Roche Holdings (RHHBY.PK) made a $44B offer for the shares of Genetech (DNA) it doesn't own. Both targets want the bid prices raised. Barron's Lawrence Strauss says these are just the two latest examples of an increasingly common biotech strategy to use mergers, buyouts, and takeovers as a way to fatten product pipeline (and profit) by acquiring promising and pre-tested biotech drugs.
Big Pharma is feeling the need to find new products with blockbuster potential as several important drugs approach the expiration of their patent protection. This, of course, will open the market to cheaper, generic versions of the drug, cutting into Big Pharma profit. Leading the pack will be Pfizer's (PFE) cholesterol drug, Lipitor, which generated more than $12B in global sales last year. Other companies with expiring blockbuster patents include Wyeth (WYE), Merck (MRK), and Eli Lilly (LLY). Jay Markowitz, a T. Rowe Price health-care analyst, notes "a number of companies are facing a significant patent cliff, where billions in revenues are going to disappear."
Many large pharmaceuticals have lots of cash on their balance sheets, making acquisitions an affordable option. The weaker dollar has also made U.S. companies look more attractive to biotech and pharmaceutical firms abroad. Another major factor is the difficult process of receiving FDA approval. This lengthy and grueling process provides an additional incentive to buy companies that have already received FDA approval on their drugs, ensuring smooth pipeline production going forward.
Potential buyout targets to keep an eye on: Amylin Pharmaceuticals (AMLN), United Therapeutics (UTHR), Alexion Pharmaceuticals (ALXN), Onyx Pharmaceuticals (ONXX), Vertex Pharmaceuticals (VRTX).
*********
Thapar and other analysts point to Onyx Pharmaceuticals (ONXX) as a potential target of Bayer. Onyx has a joint venture with the German pharmaceutical giant for Nexavar, which is used to treat kidney and liver cancers. "At some point, you could see Bayer wanting to control 100% of the assets," says Sustersic, who thinks that Bayer might pay as much as $65 per share. Nexavar is believed to have more upside, especially as it secures approval in other markets (it was recently given the green light by China as a liver-cancer treatment), and even more if it can be used for other cancers, including those of the breast, lungs and skin.
Not all of the focus is on products already in the market, however. Vertex Pharmaceuticals (VRTX), another company that some investors view as an eventual takeover candidate, is developing Telaprevir, a substance that promises to reduce the time needed to treat hepatitis-C, a potentially fatal liver ailment. Telaprevir is currently in stage III testing, the last phase of the clinical- trial process. Vertex has worked with Johnson & Johnson (JNJ) in developing that drug.
Another company that could spark an acquirer's interest is Amylin Pharmaceuticals (AMLN). It's working on a Type 2 diabetes drug that could be injected once a week instead of daily. Amylin has several development partners, including Eli Lilly, on the drug. It already markets two diabetes drugs, Symlin and Byetta. But some analysts say the product under development could boast major advantages. "A once-weekly drug that lowers glucose substantially, induces weight loss, isn't associated with hypoglycemia, and lacks a cardiovascular safety signal has multibillion-dollar potential," says Markowitz.
Anyone scanning the biotech ranks for potential acquisition targets shouldn't overlook United Therapeutics (UTHR). It has one characteristic that lots of other small biotechs would envy: It's in the black. One of its most promising products is Remodulin, which is used to treat hypertension in the blood vessels of the lungs. Although there are other PAH drugs, Turner Investment Partners' Sustersic says that United Therapeutics is "one of the clear leaders, and they potentially could have a big blockbuster."
American Century's Thapar is similarly impressed by Alexion Pharmaceuticals (ALXN), which earned six cents a share in its most recent quarter, versus a loss of 75 cents a share a year earlier. Its drugs include Soliris, which treats a rare disorder called paroxysmal nocturnal hemoglobinuria, which destroys red blood cells. At $389,000 a year per patient, Soliris is hugely expensive -- but it targets a clearly defined patient base. The drug had net sales of nearly $60 million in the second quarter, up from $45.5 million in the first quarter and $9.8 million a year earlier. Regulators have approved its use in the U.S. and Europe, and it's expected to be introduced in Japan toward the end of next year. Alexion retains distribution rights in the U.S. and overseas, says Sustersic, who maintains that Soliris sales could reach at least $500 million a year.
mfg ipollit
"Die Experten von "Global Biotech Investing" halten die Onyx Pharmaceuticals-Aktie (ISIN US6833991093/ WKN 900778) für ein interessantes Investment.
Für die Experten stelle die Biotech-Gesellschaft ein attraktives Übernahmeziel dar. Das Unternehmen habe mit Nexavar das einzige am Markt zugelassene Mittel gegen Leberkrebs. Zudem habe es im letzten Jahr die Zulassung für Nierenkrebs erhalten.
Als zu Beginn des Jahres enttäuschende Daten bei Tests gegen Lungenkrebs publiziert worden seien, sei es bei dem Titel zu einem regelrechten Sell-off gekommen. So seien zeitweise Tiefstkurse um 26 USD erreicht worden. Inzwischen habe sich das Papier auf 40 USD erholt. Doch dieses Niveau könnte noch nicht das Ende der Fahnenstange gewesen sein. So würden sich am Markt hartnäckig Gerüchte halten, dass nicht nur die aktuell im Umlauf befindlichen Umsatz- und Ertragsschätzungen für die bevorstehenden Indikationen weit unter den tatsächlichen erreichbaren Niveaus lägen, sondern dass auch der Wirkstoff selbst - trotz der Enttäuschung vom Februar - noch für einiges mehr gut sei.
Temporäre Kursschwächen sollten sich bei der Onyx Pharmaceuticals-Aktie damit für mutige und visionär agierende Investoren als höchst attraktive Einstiegschance erweisen können, so die Experten von "Global Biotech Investing". (Ausgabe 14 vom 21.07.2008)
mfg ipollit
Für die Experten stelle die Biotech-Gesellschaft ein attraktives Übernahmeziel dar. Das Unternehmen habe mit Nexavar das einzige am Markt zugelassene Mittel gegen Leberkrebs. Zudem habe es im letzten Jahr die Zulassung für Nierenkrebs erhalten.
Als zu Beginn des Jahres enttäuschende Daten bei Tests gegen Lungenkrebs publiziert worden seien, sei es bei dem Titel zu einem regelrechten Sell-off gekommen. So seien zeitweise Tiefstkurse um 26 USD erreicht worden. Inzwischen habe sich das Papier auf 40 USD erholt. Doch dieses Niveau könnte noch nicht das Ende der Fahnenstange gewesen sein. So würden sich am Markt hartnäckig Gerüchte halten, dass nicht nur die aktuell im Umlauf befindlichen Umsatz- und Ertragsschätzungen für die bevorstehenden Indikationen weit unter den tatsächlichen erreichbaren Niveaus lägen, sondern dass auch der Wirkstoff selbst - trotz der Enttäuschung vom Februar - noch für einiges mehr gut sei.
Temporäre Kursschwächen sollten sich bei der Onyx Pharmaceuticals-Aktie damit für mutige und visionär agierende Investoren als höchst attraktive Einstiegschance erweisen können, so die Experten von "Global Biotech Investing". (Ausgabe 14 vom 21.07.2008)
mfg ipollit
Onyx Pharmaceuticals (ONXX)
http://www.onyx-pharm.com/
Marktkapitalisierung: 2,44 Mrd USD
Cash: 471 Mio USD
Schulden: 25 Mio USD
Umsatz 2008e/2009e (laut yahoo): 127 / 198 Mio USD
Gewinn 2008e/2009e (laut yahoo): 20 / 63 Mio USD
zugelassene Produkte: Nexavar (Leber- und Nierenkrebs)
Partner: Bayer (Hälfte der Umsätze und Gewinne weltweit)
Pipeline:
Onyx besitzt eigentlich nur Nexavar, dass als orales Krebsmittel bereits für Leber und Nierenkrebs (bei Nierenkrebs ist Sutent von Pfizer eine großer Konkurrent) zugelassen ist. Zusätzlich wird es noch u.a. gegen Lungen-, Brust- und Hautkrebs getestet.

Leberkrebs ist eines er Krebsarten, gegen die bis jetzt wenig hilft... es ist die Krebsart mit den drittmeisten Todesfällen weltweit. Nexavar ist eins der wenigen Mittel, die bei Leberkrebs wirken:
Drug May Be Breakthrough For Liver Cancer
Matthew Herper, 06.04.07, 10:55 AM ET
CHICAGO - A crying researcher woke Gunnar Riemann, the head of Bayer's $10 billion drug business, from his bed Feb. 9. They were tears of joy: A study had found that Nexavar, a drug Bayer invented and developed with tiny biotech Onyx Pharmaceuticals of Emeryville, Calif., extended the lives of patients with advanced liver cancer, the first time a treatment has been demonstrated to be effective for the hard to treat disease.
Onyx shares jumped almost 10%, to $33.83, in early morning trading Monday.
Over the past 30 years, doctors have conducted about a 100 studies on various treatments to help these very sick patients, to no avail.
However, in the case of Nexavar, Phase III clinical trials with 602 patients showed that it extended the life of the average patient by 2.8 months to 10.7 months, as compared to patients who received a placebo. Side effects were benign for a cancer drug, with the most common being severe diarrhea and rashes on the hands and feet.
The data were set to be presented here Monday morning at the annual meeting of the American Society of Clinical Oncology.
"The results are a breakthrough," says Josep Llovet, a liver specialist with the Hospital Clinic of Barcelona and the Mount Sinai School of Medicine in New York who helped organize and conduct the trial for Bayer (nyse: BAY - news - people ). "The difference in survival is completely unprecedented. You are freezing the tumor and delaying the progression of the disease."
Liver cancer is one of the deadliest of cancers. It afflicts 19,000 people in the U.S. each year, killing the vast majority. For many patients with the advanced form of the disease, Nexavar will now become the standard treatment, eventually generating hundreds of millions in sales for the partners.
Liver cancer is far more prevalent overseas, with 600,000 new cases a year worldwide. The highest rates are in Asia, where hepatitis B and C are common. Nexavar is still being tested there.
Bayer and Onyx (nasdaq: ONXX - news - people ) have been working toward this victory for 13 years. Bayer chemists developed Nexavar based on proteins Onyx identified that showed promise for halting tumor growth. The drug has already been approved and marketed for treating kidney tumors, and the two companies last year spent $160 million testing it in new diseases.
While the promising results for Nexavar may bring tears to the eyes of Bayer researchers and hope to cancer patients, doctors have lately become used to such breakthroughs, with pharmaceutical giants like Pfizer (nyse: PFE - news - people ), GlaxoSmithKline (nyse: GSK - news - people ) and AstraZeneca (nyse: AZN - news - people ) testing dozens of new targeted cancer pills. Bayer recently bought German rival Schering AG[orgid partly to get its hands on that company's cancer drug pipeline.
This year's meeting of the American Society of Clinical Oncology (ASCO) is a "building year," says David Parkinson, head of oncology at Biogen Idec (nasdaq: BIIB - news - people ). David Schenkein, a Genentech (nyse: DNA - news - people ) vice president, says the advances are "incremental."
Pfizer's kidney cancer drug Sutent has been outpacing Nexavar, with sales of $102 million versus $60 million for Nexavar in the first quarter of 2007. But study results for Sutent on liver cancer look less promising, and an abstract already presented here seems to point to a far worse side-effect profile for Sutent than Nexavar in treating that form of the disease. Reduced white blood cell and platelet counts were common, and there were five patients who developed very serious problems, including liver problems and bleeding. Pfizer notes that the study is small and early.

besonders viele Fälle von Leberkrebs gibt es in China. Hier hat Nexavar vor Kurzen die Zulassung erhalten.

Bayer: Nexavar in China auch bei Leberkrebs genehmigt
Montag, 28. Juli 2008, 10:42 Uhr
Frankfurt (Reuters) - Der Pharmakonzern Bayer hat in China eine zweite große Zulassung für sein Krebsmittel Nexavar erhalten.
Die chinesische Arzneimittelbehörde habe dem Medikament nun auch die Zulassung zur Behandlung von fortgeschrittenem Leberkrebs erteilt, teilte Bayer am Montag in Leverkusen mit. "China hat weltweit die höchste Zahl an Patienten mit Leberkrebs. Jedes Jahr werden mehr als 340.000 Fälle diagnostiziert, und die Anzahl der Neuerkrankungen nimmt weiter zu", sagte Bayer HealthCare-Vorstand Gunnar Riemann. Zu Behandlung von Nierenkrebs hat Nexavar im Reich der Mitte bereits die Genehmigung.
Das Präparat ist einer der größten Hoffnungsträger des Bayer-Pharmageschäfts. Für alle geplanten Einsatzgebiete hat Bayer für Nexavar jährliche Spitzenumsätze von mehr als zwei Milliarden Euro in Aussicht gestellt. Im ersten Quartal hatte Bayer mit Nexavar, das zur Behandlung von Nierenkrebs bereits für mehr als 70 Länder und zur Therapie von Leberkrebs für über 40 Länder zugelassen ist, 101 Millionen Euro umgesetzt. Im Vorjahreszeitraum waren es 47 Millionen Euro. Die Leverkusener entwickeln das Krebsmittel zusammen mit der US-Firma Onyx Pharmaceuticals.
Nexavar bekämpft Krebstumore, indem es zum einen durch die Hemmung eines Enzyms das Wachstum der Tumorzellen stoppt. Außerdem greift es in die Gefäßneubildung ein, um die Blutversorgung des Tumors zu unterbinden.
Die häufigste Ursache für Leberkrebs ist nach Angaben von Bayer eine chronische Infektion mit Hepatitis-B oder Hepatitis-C. In Asien seien mehr als acht Prozent der Bevölkerung mit dem Hepatatis-B und bis zu vier Prozent mit dem Hepatitis-C-Virus infiziert.
bisher sieht die Umsatzentwicklung von Nexavar positiv aus...

Onyx Has Blockbuster Potential
Posted Wed Aug 06, 04:12 pm ET
Posted By: Grant Zeng, CFA
Onyx Pharmaceuticals Inc.’s (ONXX) second-quarter financials were a little disappointing, but we continue to believe Nexavar will perform well in the second half of 2008 and beyond. We believe sales of the drug will continue to grow over the next several years since the label has expanded to liver cancer.
The current Nexavar sales driver is apparently the liver cancer market while Nexavar’s market share for kidney cancer has stabilized. This has been evidenced by the worldwide Nexavar sales in the first half of 2008.
In addition to the US and EU, Nexavar has been approved in South Korea and China for liver cancer. Demographically Asia, especially China, is a much bigger market for liver cancer in terms of patient population. Other international filing is underway. Label expansion for Nexavar into other cancer indications such as breast cancer is under way.
We believe Nexavar has the potential to become a blockbuster for Onyx and model sales of the drug will reach $.1 billion in 2010. Current price is attractive and we recommend investor to buy its shares at current levels. We arrive at our $46 price target by using biotech industry average P/E of 38 x, multiplied by our estimated 2010 EPS of $1.73, discounted at 20% for two years.
****
Anfang des Jahres gab es einen großeren Einbruch, weil eine Lungenkrebs PIII gestoppt wurde, da sie keine Wirksamkeit von Nexavar zeigte.
Enttäuschend war zuletzt der schwache Ausblick. Die Prognose für das Gesamtjahr lässt vielleicht sogar noch Quartale mit Verlust erwarten... u.U. wegen der gestiegenen Forschungsausgaben. Langfristig sollte dies allerdings keine Rolle spielen.


mfg ipollit
http://www.onyx-pharm.com/
Marktkapitalisierung: 2,44 Mrd USD
Cash: 471 Mio USD
Schulden: 25 Mio USD
Umsatz 2008e/2009e (laut yahoo): 127 / 198 Mio USD
Gewinn 2008e/2009e (laut yahoo): 20 / 63 Mio USD
zugelassene Produkte: Nexavar (Leber- und Nierenkrebs)
Partner: Bayer (Hälfte der Umsätze und Gewinne weltweit)
Pipeline:
Onyx besitzt eigentlich nur Nexavar, dass als orales Krebsmittel bereits für Leber und Nierenkrebs (bei Nierenkrebs ist Sutent von Pfizer eine großer Konkurrent) zugelassen ist. Zusätzlich wird es noch u.a. gegen Lungen-, Brust- und Hautkrebs getestet.
Leberkrebs ist eines er Krebsarten, gegen die bis jetzt wenig hilft... es ist die Krebsart mit den drittmeisten Todesfällen weltweit. Nexavar ist eins der wenigen Mittel, die bei Leberkrebs wirken:
Drug May Be Breakthrough For Liver Cancer
Matthew Herper, 06.04.07, 10:55 AM ET
CHICAGO - A crying researcher woke Gunnar Riemann, the head of Bayer's $10 billion drug business, from his bed Feb. 9. They were tears of joy: A study had found that Nexavar, a drug Bayer invented and developed with tiny biotech Onyx Pharmaceuticals of Emeryville, Calif., extended the lives of patients with advanced liver cancer, the first time a treatment has been demonstrated to be effective for the hard to treat disease.
Onyx shares jumped almost 10%, to $33.83, in early morning trading Monday.
Over the past 30 years, doctors have conducted about a 100 studies on various treatments to help these very sick patients, to no avail.
However, in the case of Nexavar, Phase III clinical trials with 602 patients showed that it extended the life of the average patient by 2.8 months to 10.7 months, as compared to patients who received a placebo. Side effects were benign for a cancer drug, with the most common being severe diarrhea and rashes on the hands and feet.
The data were set to be presented here Monday morning at the annual meeting of the American Society of Clinical Oncology.
"The results are a breakthrough," says Josep Llovet, a liver specialist with the Hospital Clinic of Barcelona and the Mount Sinai School of Medicine in New York who helped organize and conduct the trial for Bayer (nyse: BAY - news - people ). "The difference in survival is completely unprecedented. You are freezing the tumor and delaying the progression of the disease."
Liver cancer is one of the deadliest of cancers. It afflicts 19,000 people in the U.S. each year, killing the vast majority. For many patients with the advanced form of the disease, Nexavar will now become the standard treatment, eventually generating hundreds of millions in sales for the partners.
Liver cancer is far more prevalent overseas, with 600,000 new cases a year worldwide. The highest rates are in Asia, where hepatitis B and C are common. Nexavar is still being tested there.
Bayer and Onyx (nasdaq: ONXX - news - people ) have been working toward this victory for 13 years. Bayer chemists developed Nexavar based on proteins Onyx identified that showed promise for halting tumor growth. The drug has already been approved and marketed for treating kidney tumors, and the two companies last year spent $160 million testing it in new diseases.
While the promising results for Nexavar may bring tears to the eyes of Bayer researchers and hope to cancer patients, doctors have lately become used to such breakthroughs, with pharmaceutical giants like Pfizer (nyse: PFE - news - people ), GlaxoSmithKline (nyse: GSK - news - people ) and AstraZeneca (nyse: AZN - news - people ) testing dozens of new targeted cancer pills. Bayer recently bought German rival Schering AG[orgid partly to get its hands on that company's cancer drug pipeline.
This year's meeting of the American Society of Clinical Oncology (ASCO) is a "building year," says David Parkinson, head of oncology at Biogen Idec (nasdaq: BIIB - news - people ). David Schenkein, a Genentech (nyse: DNA - news - people ) vice president, says the advances are "incremental."
Pfizer's kidney cancer drug Sutent has been outpacing Nexavar, with sales of $102 million versus $60 million for Nexavar in the first quarter of 2007. But study results for Sutent on liver cancer look less promising, and an abstract already presented here seems to point to a far worse side-effect profile for Sutent than Nexavar in treating that form of the disease. Reduced white blood cell and platelet counts were common, and there were five patients who developed very serious problems, including liver problems and bleeding. Pfizer notes that the study is small and early.
besonders viele Fälle von Leberkrebs gibt es in China. Hier hat Nexavar vor Kurzen die Zulassung erhalten.
Bayer: Nexavar in China auch bei Leberkrebs genehmigt
Montag, 28. Juli 2008, 10:42 Uhr
Frankfurt (Reuters) - Der Pharmakonzern Bayer hat in China eine zweite große Zulassung für sein Krebsmittel Nexavar erhalten.
Die chinesische Arzneimittelbehörde habe dem Medikament nun auch die Zulassung zur Behandlung von fortgeschrittenem Leberkrebs erteilt, teilte Bayer am Montag in Leverkusen mit. "China hat weltweit die höchste Zahl an Patienten mit Leberkrebs. Jedes Jahr werden mehr als 340.000 Fälle diagnostiziert, und die Anzahl der Neuerkrankungen nimmt weiter zu", sagte Bayer HealthCare-Vorstand Gunnar Riemann. Zu Behandlung von Nierenkrebs hat Nexavar im Reich der Mitte bereits die Genehmigung.
Das Präparat ist einer der größten Hoffnungsträger des Bayer-Pharmageschäfts. Für alle geplanten Einsatzgebiete hat Bayer für Nexavar jährliche Spitzenumsätze von mehr als zwei Milliarden Euro in Aussicht gestellt. Im ersten Quartal hatte Bayer mit Nexavar, das zur Behandlung von Nierenkrebs bereits für mehr als 70 Länder und zur Therapie von Leberkrebs für über 40 Länder zugelassen ist, 101 Millionen Euro umgesetzt. Im Vorjahreszeitraum waren es 47 Millionen Euro. Die Leverkusener entwickeln das Krebsmittel zusammen mit der US-Firma Onyx Pharmaceuticals.
Nexavar bekämpft Krebstumore, indem es zum einen durch die Hemmung eines Enzyms das Wachstum der Tumorzellen stoppt. Außerdem greift es in die Gefäßneubildung ein, um die Blutversorgung des Tumors zu unterbinden.
Die häufigste Ursache für Leberkrebs ist nach Angaben von Bayer eine chronische Infektion mit Hepatitis-B oder Hepatitis-C. In Asien seien mehr als acht Prozent der Bevölkerung mit dem Hepatatis-B und bis zu vier Prozent mit dem Hepatitis-C-Virus infiziert.
bisher sieht die Umsatzentwicklung von Nexavar positiv aus...
Onyx Has Blockbuster Potential
Posted Wed Aug 06, 04:12 pm ET
Posted By: Grant Zeng, CFA
Onyx Pharmaceuticals Inc.’s (ONXX) second-quarter financials were a little disappointing, but we continue to believe Nexavar will perform well in the second half of 2008 and beyond. We believe sales of the drug will continue to grow over the next several years since the label has expanded to liver cancer.
The current Nexavar sales driver is apparently the liver cancer market while Nexavar’s market share for kidney cancer has stabilized. This has been evidenced by the worldwide Nexavar sales in the first half of 2008.
In addition to the US and EU, Nexavar has been approved in South Korea and China for liver cancer. Demographically Asia, especially China, is a much bigger market for liver cancer in terms of patient population. Other international filing is underway. Label expansion for Nexavar into other cancer indications such as breast cancer is under way.
We believe Nexavar has the potential to become a blockbuster for Onyx and model sales of the drug will reach $.1 billion in 2010. Current price is attractive and we recommend investor to buy its shares at current levels. We arrive at our $46 price target by using biotech industry average P/E of 38 x, multiplied by our estimated 2010 EPS of $1.73, discounted at 20% for two years.
****
Anfang des Jahres gab es einen großeren Einbruch, weil eine Lungenkrebs PIII gestoppt wurde, da sie keine Wirksamkeit von Nexavar zeigte.
Enttäuschend war zuletzt der schwache Ausblick. Die Prognose für das Gesamtjahr lässt vielleicht sogar noch Quartale mit Verlust erwarten... u.U. wegen der gestiegenen Forschungsausgaben. Langfristig sollte dies allerdings keine Rolle spielen.
mfg ipollit
Antwort auf Beitrag Nr.: 34.745.188 von ipollit am 17.08.08 20:53:13http://www.fool.com/investing/general/2008/08/06/onyx-in-the…
Onyx in the Black -- for Now
By Brian Orelli
August 6, 2008
For investors in Onyx Pharmaceuticals (Nasdaq: ONXX), there was no question as to what the top-line sales of its only drug were going to be; partner Bayer spilled the beans last week. The big question last night was whether it kept costs under control. The stock is in the red today, so you can guess what investors thought of the bottom line.
Sales of the duo's cancer treatment, Nexavar, more than doubled from the second quarter of last year to more than $168 million. Most of that increase is attributable to the launch of Nexavar as a treatment for liver cancer, where the drug pretty much has the market all to itself. Nexavar is also approved for treating kidney disease, but competes heavily for patients with Pfizer's (NYSE: PFE) Sutent.
On the bottom line, Onyx brought in $0.08 per share. Anything that's positive is usually a good sign for a small drug company, but that's less than a third of what it brought in during the first quarter on lower drug sales. At one time, analysts expected full-year earnings of $0.91 per share. Currently, the expectation is $0.58. Based on management's latest comments, that probably won't happen.
On the earnings conference call, Onyx said it expects to be "breakeven or profitable on the bottom line for the full year 2008." Being conservative and doing the math, that means investors should expect some red ahead as the company spends more on marketing, as well as on clinical trials to get Nexavar approved for other indications.
A quick search of clinicaltrials.gov shows dozens and dozens of trials. Now, granted, Onyx and Bayer aren't paying for all of the clinical trials Nexavar is involved in, but they are funding quite a few, including using Nexavar in combination with already approved medications like Eli Lilly's (NYSE: LLY) Alimta or Tarceva from OSI Pharmaceuticals (Nasdaq: OSIP) and Genentech (NYSE: DNA).
I'm not sure whether it's the fact that Nexavar failed to show an effect in lung cancer earlier this year or because Onyx has a new CEO running the show, but the one-drug-wonder appears to be shying away from its mantra that Nexavar is a "pipeline in a drug." Instead, it said that it expects to expand its pipeline in the future -- presumably through acquisitions, although this wasn't explicitly stated.
That's always been my issue with Onyx. Nexavar may be a wonder drug, but without a pipeline to speak of, the company's long-term future has always been in question. The slight shift in focus has piqued my interest, but I'll be sitting on the sidelines until we know exactly what the company has in store for its next big thing.
mfg ipollit
Onyx in the Black -- for Now
By Brian Orelli
August 6, 2008
For investors in Onyx Pharmaceuticals (Nasdaq: ONXX), there was no question as to what the top-line sales of its only drug were going to be; partner Bayer spilled the beans last week. The big question last night was whether it kept costs under control. The stock is in the red today, so you can guess what investors thought of the bottom line.
Sales of the duo's cancer treatment, Nexavar, more than doubled from the second quarter of last year to more than $168 million. Most of that increase is attributable to the launch of Nexavar as a treatment for liver cancer, where the drug pretty much has the market all to itself. Nexavar is also approved for treating kidney disease, but competes heavily for patients with Pfizer's (NYSE: PFE) Sutent.
On the bottom line, Onyx brought in $0.08 per share. Anything that's positive is usually a good sign for a small drug company, but that's less than a third of what it brought in during the first quarter on lower drug sales. At one time, analysts expected full-year earnings of $0.91 per share. Currently, the expectation is $0.58. Based on management's latest comments, that probably won't happen.
On the earnings conference call, Onyx said it expects to be "breakeven or profitable on the bottom line for the full year 2008." Being conservative and doing the math, that means investors should expect some red ahead as the company spends more on marketing, as well as on clinical trials to get Nexavar approved for other indications.
A quick search of clinicaltrials.gov shows dozens and dozens of trials. Now, granted, Onyx and Bayer aren't paying for all of the clinical trials Nexavar is involved in, but they are funding quite a few, including using Nexavar in combination with already approved medications like Eli Lilly's (NYSE: LLY) Alimta or Tarceva from OSI Pharmaceuticals (Nasdaq: OSIP) and Genentech (NYSE: DNA).
I'm not sure whether it's the fact that Nexavar failed to show an effect in lung cancer earlier this year or because Onyx has a new CEO running the show, but the one-drug-wonder appears to be shying away from its mantra that Nexavar is a "pipeline in a drug." Instead, it said that it expects to expand its pipeline in the future -- presumably through acquisitions, although this wasn't explicitly stated.
That's always been my issue with Onyx. Nexavar may be a wonder drug, but without a pipeline to speak of, the company's long-term future has always been in question. The slight shift in focus has piqued my interest, but I'll be sitting on the sidelines until we know exactly what the company has in store for its next big thing.
mfg ipollit
http://www.godmode-trader.de/de/aktie-analyse/Amex-Biotech-I…
Amex Biotech Index - Unschlagbar stark - Neue AllTimeHighs
Datum 15.08.2008 - Uhrzeit 17:09
Kursstand: 888,59 Punkte
Rückblick: Der Amex Biotech Index vollzog in den vergangenen Jahren eine deutliche Aufwärtsbewegung und markierte ein neues AllTimeHigh im Oktober 2007 bei 864,82 Punkten. Dort startete eine Zwischenkorrektur bis an den Unterstützungsbereich aus Aufwärtstrend und EMA200 auf Wochenbasis bei 670,00 - 679,00 Punkten, wo der Kurssturz im März abgefangen wurde.
Nach einer mehrmonatigen, lethargischen Seitwärtsbewegung startete im Juli eine hochdynamische Rallyebewegung, welche zum Anstieg bis ans AllTimeHigh führte. Ein erster Ausbruchsversuch scheiterte, eine Seitwärtskonsolidierung unterhalb davon wird in dieser Woche nach oben hin aufgelöst. Das Chartbild ist auf sämtlichen Zeitebenen bullisch zu werten.
Charttechnischer Ausblick: Der Amex Biotech Index markiert heute ein neues AllTimeHigh bei 893,89 Punkten und könnte als Stärkster der US Sektorenindizes die laufende Rallyebewegung in den kommenden Monaten noch weiter bis zunächst 940,00 - 950,00 Punkte fortsetzen.
Kurzfristige Rücksetzer sollten idealerweise bei 863,00 - 875,00 Punkten enden. Geht es wieder signifikant unter 855,00 Punkte, wird eine Abwärtskorrektur bis 813,00 - 820,166 Punkte und darunter ggf. sogar 787,97 Punkte möglich.

mfg ipollit
Amex Biotech Index - Unschlagbar stark - Neue AllTimeHighs
Datum 15.08.2008 - Uhrzeit 17:09
Kursstand: 888,59 Punkte
Rückblick: Der Amex Biotech Index vollzog in den vergangenen Jahren eine deutliche Aufwärtsbewegung und markierte ein neues AllTimeHigh im Oktober 2007 bei 864,82 Punkten. Dort startete eine Zwischenkorrektur bis an den Unterstützungsbereich aus Aufwärtstrend und EMA200 auf Wochenbasis bei 670,00 - 679,00 Punkten, wo der Kurssturz im März abgefangen wurde.
Nach einer mehrmonatigen, lethargischen Seitwärtsbewegung startete im Juli eine hochdynamische Rallyebewegung, welche zum Anstieg bis ans AllTimeHigh führte. Ein erster Ausbruchsversuch scheiterte, eine Seitwärtskonsolidierung unterhalb davon wird in dieser Woche nach oben hin aufgelöst. Das Chartbild ist auf sämtlichen Zeitebenen bullisch zu werten.
Charttechnischer Ausblick: Der Amex Biotech Index markiert heute ein neues AllTimeHigh bei 893,89 Punkten und könnte als Stärkster der US Sektorenindizes die laufende Rallyebewegung in den kommenden Monaten noch weiter bis zunächst 940,00 - 950,00 Punkte fortsetzen.
Kurzfristige Rücksetzer sollten idealerweise bei 863,00 - 875,00 Punkten enden. Geht es wieder signifikant unter 855,00 Punkte, wird eine Abwärtskorrektur bis 813,00 - 820,166 Punkte und darunter ggf. sogar 787,97 Punkte möglich.
mfg ipollit
Antwort auf Beitrag Nr.: 34.639.110 von ipollit am 03.08.08 20:18:25habe aus Zeitgründen noch nicht alles gelesen,
finde aber, daß man von dir eine Menge lernen kann.
finde aber, daß man von dir eine Menge lernen kann.
Hi Ipollit,
vielen Dank für die Super Beiträge speziell zu ONYX.
Ich bin da ja auch schon einige Jahre investiert und ich denke
bzw. hoffe immer noch, das ich mal den Kurs Dreistellig sehen werde.
Was ist denn deiner Meinung nach in den nächsten 2 Jahren für ONYX drin?
Merci und Gruß ONYX
vielen Dank für die Super Beiträge speziell zu ONYX.
Ich bin da ja auch schon einige Jahre investiert und ich denke
bzw. hoffe immer noch, das ich mal den Kurs Dreistellig sehen werde.
Was ist denn deiner Meinung nach in den nächsten 2 Jahren für ONYX drin?
Merci und Gruß ONYX

Antwort auf Beitrag Nr.: 34.753.053 von ONYXLeader am 18.08.08 20:42:40theoretisch sind 3-stellige Kurse möglich... ein konkrete Vorstellung habe ich vom weiteren Verlauf allerdings nicht. Ich lass mich mal überraschen, Potential ist jedenfalls vorhanden.
mfg ipollit
mfg ipollit
zu Cubist (CBST)...
[ULR]http://www.boston.com/business/healthcare/articles/2008/08/1…[/url]
For Cubist, a tough sell
Despite robust US sales of its antibiotic Cubicin, the Lexington biotech finds global success is difficult to achieve
Since Cubist Pharmaceuticals Inc. launched its first drug five years ago - an antibiotic called Cubicin - sales have skyrocketed.
This year alone, the Lexington biotech company expects Cubicin to generate $395 million to $405 million in sales nationwide, up more than one-quarter from 2007. And executives predict that US sales could climb to $750 million within the next few years. Domestic sales have been so strong that the company next month will add 187,000 square feet to its Lexington headquarters.
But sales have been tepid elsewhere in the world. Cubist expects to reap only about $7 million in royalties this year from Cubicin sales outside the United States. In documents filed with the Securities and Exchange Commission, Cubicin acknowledged that European sales have been disappointing.
"People have been asking why their international sales haven't been stronger," said Howard Liang, a biotech analyst with the Boston investment bank Leerink Swann & Co.
The problems illustrate the challenge US drug companies sometimes face when marketing drugs in other parts of the world, where they must navigate through different healthcare and regulatory systems. One analyst said drug companies need to learn to customize their approach for each country, because every one is different.
"That's a hard message for pharmaceuticals companies," said Todd Evans, who leads PricewaterhouseCoopers's commercial life science practice. "Your pricing strategy needs to be quite different. Your IP protections vary. You have different attitudes for different payers," referring to insurers and other entities that ultimately pay for the drugs.
But several Massachusetts companies - including two Cambridge biotechs - have found success overseas. Slightly more than half of Genzyme Corp.'s product revenue now comes from outside the United States. And Biogen Idec Inc. expects foreign sales to account for two-fifths of its revenue by 2010.
International sales are also key to Cubist's long-term growth plans. The company hopes Cubicin will eventually become a blockbuster - generating $1 billion or more in annual sales, including hundreds of millions of dollars from outside the country.
The drug, also called Daptomycin, was discovered by Eli Lilly & Co. researchers in the early 1980s. But Eli Lilly halted work on it because of concern about possible side effects. Cubist subsequently licensed the rights to it in 1997, spent several years on further research and development, and ultimately won approval from the Food and Drug Administration to market the drug on Sept. 12, 2003. (Cubist plans to hold an anniversary celebration next month.)
Cubicin is mainly used to fight skin and infections caused by certain bacteria, including methicillin-resistant Staphylococcus aureus, commonly referred to as MRSA. Unlike many more common antibiotics, Cubicin is administered intravenously instead of with a pill or capsule. But because it needs to be administered only once a day, Cubist has been able to tap the home market and hospitals, boosting sales.
Still, Cubist's chief executive, Michael Bonney, said Cubicin has been slower to catch on in Europe than in the United States for several reasons. It took three years longer to get approval to sell the drug in the European Union, and Cubicin still needs approval in some other parts of the world. Also, Cubicin faces additional competition abroad. And it's possible the market for the drug could be larger in the United States than in most other countries.
Nonetheless, Cubist has been disappointed with sales by its European partner, Novartis AG. Cubist signed a deal with Chiron to market the drug in Europe in 2003. But shortly afterward, Chiron was sold to Novartis, putting the drug in limbo as the two companies worked on how to combine their operations.
Then, even after Novartis decided to go ahead with Cubicin, Bonney said, the sales pitch sometimes fell flat.
"Neither we nor Novartis are satisfied with the results," said Bonney.
Cubicin's foreign sales are somewhat understated because Cubist reports only its royalties from overseas sales - thought to be one-third of the drug's total foreign revenue. (Novartis and other partners keep the rest.) But even a tripling of Cubicin's foreign revenue would amount only to $21 million this year, about 5 percent of the drug's worldwide revenue.
Bonney said Novartis made a tactical error: trying to market the drug to every hospital with the same marketing pitch, instead of tailoring its message. But he said Novartis and Cubist are now working together to refine how the drug is presented to potential buyers.
Their effort may be paying off - Cubist recently said it saw strong sales growth for Cubicin in certain countries, including the United Kingdom and Germany.
"Outside the US, we see signs of progress," chief operating officer Rob Perez told investors last month.
A Novartis spokeswoman said the Swiss drug maker was happy with its results, despite Cubist's past complaints. She said the drug was on track to meet its goals this year.
mfg ipollit
[ULR]http://www.boston.com/business/healthcare/articles/2008/08/1…[/url]
For Cubist, a tough sell
Despite robust US sales of its antibiotic Cubicin, the Lexington biotech finds global success is difficult to achieve
Since Cubist Pharmaceuticals Inc. launched its first drug five years ago - an antibiotic called Cubicin - sales have skyrocketed.
This year alone, the Lexington biotech company expects Cubicin to generate $395 million to $405 million in sales nationwide, up more than one-quarter from 2007. And executives predict that US sales could climb to $750 million within the next few years. Domestic sales have been so strong that the company next month will add 187,000 square feet to its Lexington headquarters.
But sales have been tepid elsewhere in the world. Cubist expects to reap only about $7 million in royalties this year from Cubicin sales outside the United States. In documents filed with the Securities and Exchange Commission, Cubicin acknowledged that European sales have been disappointing.
"People have been asking why their international sales haven't been stronger," said Howard Liang, a biotech analyst with the Boston investment bank Leerink Swann & Co.
The problems illustrate the challenge US drug companies sometimes face when marketing drugs in other parts of the world, where they must navigate through different healthcare and regulatory systems. One analyst said drug companies need to learn to customize their approach for each country, because every one is different.
"That's a hard message for pharmaceuticals companies," said Todd Evans, who leads PricewaterhouseCoopers's commercial life science practice. "Your pricing strategy needs to be quite different. Your IP protections vary. You have different attitudes for different payers," referring to insurers and other entities that ultimately pay for the drugs.
But several Massachusetts companies - including two Cambridge biotechs - have found success overseas. Slightly more than half of Genzyme Corp.'s product revenue now comes from outside the United States. And Biogen Idec Inc. expects foreign sales to account for two-fifths of its revenue by 2010.
International sales are also key to Cubist's long-term growth plans. The company hopes Cubicin will eventually become a blockbuster - generating $1 billion or more in annual sales, including hundreds of millions of dollars from outside the country.
The drug, also called Daptomycin, was discovered by Eli Lilly & Co. researchers in the early 1980s. But Eli Lilly halted work on it because of concern about possible side effects. Cubist subsequently licensed the rights to it in 1997, spent several years on further research and development, and ultimately won approval from the Food and Drug Administration to market the drug on Sept. 12, 2003. (Cubist plans to hold an anniversary celebration next month.)
Cubicin is mainly used to fight skin and infections caused by certain bacteria, including methicillin-resistant Staphylococcus aureus, commonly referred to as MRSA. Unlike many more common antibiotics, Cubicin is administered intravenously instead of with a pill or capsule. But because it needs to be administered only once a day, Cubist has been able to tap the home market and hospitals, boosting sales.
Still, Cubist's chief executive, Michael Bonney, said Cubicin has been slower to catch on in Europe than in the United States for several reasons. It took three years longer to get approval to sell the drug in the European Union, and Cubicin still needs approval in some other parts of the world. Also, Cubicin faces additional competition abroad. And it's possible the market for the drug could be larger in the United States than in most other countries.
Nonetheless, Cubist has been disappointed with sales by its European partner, Novartis AG. Cubist signed a deal with Chiron to market the drug in Europe in 2003. But shortly afterward, Chiron was sold to Novartis, putting the drug in limbo as the two companies worked on how to combine their operations.
Then, even after Novartis decided to go ahead with Cubicin, Bonney said, the sales pitch sometimes fell flat.
"Neither we nor Novartis are satisfied with the results," said Bonney.
Cubicin's foreign sales are somewhat understated because Cubist reports only its royalties from overseas sales - thought to be one-third of the drug's total foreign revenue. (Novartis and other partners keep the rest.) But even a tripling of Cubicin's foreign revenue would amount only to $21 million this year, about 5 percent of the drug's worldwide revenue.
Bonney said Novartis made a tactical error: trying to market the drug to every hospital with the same marketing pitch, instead of tailoring its message. But he said Novartis and Cubist are now working together to refine how the drug is presented to potential buyers.
Their effort may be paying off - Cubist recently said it saw strong sales growth for Cubicin in certain countries, including the United Kingdom and Germany.
"Outside the US, we see signs of progress," chief operating officer Rob Perez told investors last month.
A Novartis spokeswoman said the Swiss drug maker was happy with its results, despite Cubist's past complaints. She said the drug was on track to meet its goals this year.
mfg ipollit
zu VPHM und CBST...
Behring's Berinert mögliche FDA-Zulassung scheint um ein paar Monate verschoben worden zu sein. Dies ist sehr positiv für Viropharma's Cinryze, da dies nun wieder als erstes die FDA-Zulassung für HAE erhalten kann (14. Oktober). Cinryze Vorteil liegt in einer mögliche prohylaktischen Anwendung. Dyax DX-88 oder Jerini's Icatibant kann dagegen auch subkutan angewendet werden und ist für akute Anfälle vielleicht geeigneter.
DX-88 wird nicht nur gegen die sehr seltene Krankheit HAE entwickelt, sondern auch gegen Blutverlust bei OPs... hier könnte es den vom Markt genommenen potentiellen Blockbuster Trasylol von Bayer (zu Trasylol... Trasylol wird eingesetzt, um bei Herzoperationen Blutungen zu verringern. "Das ist ein 600-Millionen-Dollar-pro-Jahr-Medikament. Es hätte ein Blockbuster für Bayer sein können", sagte Mangano. Bayer hatte die Erwartung für den Spitzenumsatz mit Trasylol kürzlich auf über 500 Millionen Euro angehoben.) Dafür hat Cubist vor einiger Zeit die US-Rechte gekauft. Die Entwicklung befindet sich allerdings erst in der PII.
http://www.thestreet.com/print/story/10433760.html
Positive Study Drives Dyax Higher
2008-08-18 14:44:54.0, DYAX SHPGY VPHM, Elizabeth Trotta
Dyax(DYAX - Cramer's Take - Stockpickr) shares shot higher Monday after the company announced positive late-stage trial results for its Hereditary Angiodema (HAE) drug DX-88, and will now complete its filing for FDA approval.
Dyax shares were up more than 10% to $4.78 in recent trading.
The company said that a phase III trial, dubbed Edema 4, successfully met its primary and secondary endpoints, replicating results from an earlier trial. Dyax also said the drug was well tolerated with no drug-related serious adverse events reported.
HAE causes periodic, acute episodes of painful swelling in a patient's extremities, gastrointestinal tract and, most dangerously, the airway passages. It's estimated that about 30% of HAE patients die due to suffocation caused when their airway swells shut.
The disease affects roughly 10,000 patients in North America, according to Dyax.
Cowen analyst Phil Nadeau, who has an outperform rating for the stock, said in a note to investors that his model projects $130 million in 2012 sales from DX-88 (Dyax posted revenue in its most recent quarter of $3.8 million, along with a loss of nearly $25 million)
With its first marketed drug, Dyax will enter a competitive market -- one that has seen a spate of regulatory delays -- for a rare disease that affects only 10,000 people in North America.
But Dyax's offering has its advantages.
DX-88 can be given to patients with a simple injection under the skin, instead of intravenously, which may make it more convenient and stronger competitor when launched.
Dyax is expected to file the last module of its application for DX-88 for FDA approval early in the fourth quarter based on the phase III study results announced on Monday. This means the company will likely have the product in market around mid-year 2009, writes Needham analyst Mark Monane, who has a buy rating and an $8 price target for the stock.
The company is in an exclusive negotiation period with Dompe regarding European rights to the DX-88 and has teamed up with Cubist(CBST - Cramer's Take - Stockpickr) for North American rights to the drug in another indication.
Other HAE drug developers have won premium takeout prices in recent months.
In July, Shire(SHPGY - Cramer's Take - Stockpickr) announced that it was buying Jerini and its lead product HAE drug Firazyr in a deal worth $521 million. However, Wall Street widely expects U.S. approval will require another clinical trial, giving Dyax a buffer (pending approval) before this competitor enters the market. ViroPharma(VPHM - Cramer's Take - Stockpickr) also announced in July that it would spend at least $443 million to buy Lev Pharmaceuticals, which is expecting to hear from the FDA on Cinryze, its intravenous HAE treatment seeking approval to treat and prevent attacks, on or before Oct. 14.
If approved, Cinryze could still compete with Behring's Berinert P, a prospective treatment seeking approval for acute HAE attacks -- not a prophylactic, or preventive treatment for the disease.
The FDA was expected to make its decision on Berinert P on or before Sept. 6, but the company confirmed with Wall Street analysts on Friday that it has been delayed (60-90 days), based on further "data slicing requirements" according to Susquehanna analyst Jason Kolbert.
Berinert P, like Cinryze, is given intravenously, not subcutaneously like DX-88 and Firazyr.
"From our conversations with HAE patients, we know that they prefer subcutaneous therapy," says Kolbert who has concerns regarding the market size estimates represented by consensus. Kolbert believes the bigger market opportunity for DX-88 is not in HAE, but in areas such as drug-induced edema and prevention of blood loss in surgery.
Dyax recently signed an agreement with Cubist for North American rights to DX-88 in blood loss prevention in heart surgery.
mfg ipollit
Behring's Berinert mögliche FDA-Zulassung scheint um ein paar Monate verschoben worden zu sein. Dies ist sehr positiv für Viropharma's Cinryze, da dies nun wieder als erstes die FDA-Zulassung für HAE erhalten kann (14. Oktober). Cinryze Vorteil liegt in einer mögliche prohylaktischen Anwendung. Dyax DX-88 oder Jerini's Icatibant kann dagegen auch subkutan angewendet werden und ist für akute Anfälle vielleicht geeigneter.
DX-88 wird nicht nur gegen die sehr seltene Krankheit HAE entwickelt, sondern auch gegen Blutverlust bei OPs... hier könnte es den vom Markt genommenen potentiellen Blockbuster Trasylol von Bayer (zu Trasylol... Trasylol wird eingesetzt, um bei Herzoperationen Blutungen zu verringern. "Das ist ein 600-Millionen-Dollar-pro-Jahr-Medikament. Es hätte ein Blockbuster für Bayer sein können", sagte Mangano. Bayer hatte die Erwartung für den Spitzenumsatz mit Trasylol kürzlich auf über 500 Millionen Euro angehoben.) Dafür hat Cubist vor einiger Zeit die US-Rechte gekauft. Die Entwicklung befindet sich allerdings erst in der PII.
http://www.thestreet.com/print/story/10433760.html
Positive Study Drives Dyax Higher
2008-08-18 14:44:54.0, DYAX SHPGY VPHM, Elizabeth Trotta
Dyax(DYAX - Cramer's Take - Stockpickr) shares shot higher Monday after the company announced positive late-stage trial results for its Hereditary Angiodema (HAE) drug DX-88, and will now complete its filing for FDA approval.
Dyax shares were up more than 10% to $4.78 in recent trading.
The company said that a phase III trial, dubbed Edema 4, successfully met its primary and secondary endpoints, replicating results from an earlier trial. Dyax also said the drug was well tolerated with no drug-related serious adverse events reported.
HAE causes periodic, acute episodes of painful swelling in a patient's extremities, gastrointestinal tract and, most dangerously, the airway passages. It's estimated that about 30% of HAE patients die due to suffocation caused when their airway swells shut.
The disease affects roughly 10,000 patients in North America, according to Dyax.
Cowen analyst Phil Nadeau, who has an outperform rating for the stock, said in a note to investors that his model projects $130 million in 2012 sales from DX-88 (Dyax posted revenue in its most recent quarter of $3.8 million, along with a loss of nearly $25 million)
With its first marketed drug, Dyax will enter a competitive market -- one that has seen a spate of regulatory delays -- for a rare disease that affects only 10,000 people in North America.
But Dyax's offering has its advantages.
DX-88 can be given to patients with a simple injection under the skin, instead of intravenously, which may make it more convenient and stronger competitor when launched.
Dyax is expected to file the last module of its application for DX-88 for FDA approval early in the fourth quarter based on the phase III study results announced on Monday. This means the company will likely have the product in market around mid-year 2009, writes Needham analyst Mark Monane, who has a buy rating and an $8 price target for the stock.
The company is in an exclusive negotiation period with Dompe regarding European rights to the DX-88 and has teamed up with Cubist(CBST - Cramer's Take - Stockpickr) for North American rights to the drug in another indication.
Other HAE drug developers have won premium takeout prices in recent months.
In July, Shire(SHPGY - Cramer's Take - Stockpickr) announced that it was buying Jerini and its lead product HAE drug Firazyr in a deal worth $521 million. However, Wall Street widely expects U.S. approval will require another clinical trial, giving Dyax a buffer (pending approval) before this competitor enters the market. ViroPharma(VPHM - Cramer's Take - Stockpickr) also announced in July that it would spend at least $443 million to buy Lev Pharmaceuticals, which is expecting to hear from the FDA on Cinryze, its intravenous HAE treatment seeking approval to treat and prevent attacks, on or before Oct. 14.
If approved, Cinryze could still compete with Behring's Berinert P, a prospective treatment seeking approval for acute HAE attacks -- not a prophylactic, or preventive treatment for the disease.
The FDA was expected to make its decision on Berinert P on or before Sept. 6, but the company confirmed with Wall Street analysts on Friday that it has been delayed (60-90 days), based on further "data slicing requirements" according to Susquehanna analyst Jason Kolbert.
Berinert P, like Cinryze, is given intravenously, not subcutaneously like DX-88 and Firazyr.
"From our conversations with HAE patients, we know that they prefer subcutaneous therapy," says Kolbert who has concerns regarding the market size estimates represented by consensus. Kolbert believes the bigger market opportunity for DX-88 is not in HAE, but in areas such as drug-induced edema and prevention of blood loss in surgery.
Dyax recently signed an agreement with Cubist for North American rights to DX-88 in blood loss prevention in heart surgery.
mfg ipollit
Evotec...
Das ist ja nichts neues. Mal sehen, ob da bald mal etwas konkretes passiert. Zuletzt klang es bei der Veröffentlichung der Quartalszahlen nicht so optimistisch: Entsprechende Gespräche für unser Produkt EVT 201 zur Behandlung von Schlafstörungen laufen. Schlaflosigkeit bleibt hinsichtlich des Abschlusses neuer Kooperationen ein schwieriges Indikationsgebiet, insbesondere nach den jüngsten Rückschlägen bei den Konkurrenz-Präparaten Gaboxadol, Indiplon und anderen. Dass der Markt aber weiterhin an diesem Indikationsgebiet Interesse zeigt, beweist der von Actelion im Juli verkündete Vertrag mit GSK zur gemeinsamen Entwicklung ihres Schlafmittels Almorexant. Erfolg in der Auslizenzierung ist für uns von enormer Bedeutung, um aus unseren Forschungsprojekten Wertschöpfung zu erzielen."
http://www.cash.ch/news/story/448/586933/40/40
Biotechfirma Evotec sucht nach finanzkräftigem Partner
19.08 09:08
HAMBURG (AWP International) - Das Biotech-Unternehmen Evotec sucht für die Entwicklung von Arzneien einen finanzkräftigen Partner aus der Pharmaindustrie. So will die Gesellschaft bis Jahresende für das am weitesten entwickelte Medikament, ein potenzielles Schlafmittel, einen "schlagkräftigen" Pharmakonzern als Partner gewonnen haben, berichtet die "Süddeutsche Zeitung" (SZ/Dienstagausgabe) unter Berufung auf Evotec-Chef Jörn Aldag. Das sei das wichtigste Jahresziel, denn Evotec plane 2009 den Start der dritten vorgeschriebenen Testphase. Das Investmenthaus Sal. Oppenheim traut dem Schlafmittel nach Zulassung Jahreserlöse in Höhe von bis zu 890 Millionen Euro zu.
Zudem forsche Evotec an einem Mittel zur Raucherentwöhnung, hiess es weiter. Dieses stecke seit Februar in Testphase II. Die Studie soll den Einfluss des Präparats auf das Suchtverhalten und die Entzugserscheinungen an 90 Rauchern zeigen. Für weitere kostspielige Tests sind die Hamburger ebenfalls auf Partner-Suche aus der Industrie. Insgesamt plant die Firma, dieses Jahr zwischen 46 und 51 Millionen Euro in die Forschung zu stecken. Die diesjährigen Umsätze dürften bei 30 bis 35 Millionen Euro liegen, sagte Aldag der Zeitung.
mfg ipollit
Das ist ja nichts neues. Mal sehen, ob da bald mal etwas konkretes passiert. Zuletzt klang es bei der Veröffentlichung der Quartalszahlen nicht so optimistisch: Entsprechende Gespräche für unser Produkt EVT 201 zur Behandlung von Schlafstörungen laufen. Schlaflosigkeit bleibt hinsichtlich des Abschlusses neuer Kooperationen ein schwieriges Indikationsgebiet, insbesondere nach den jüngsten Rückschlägen bei den Konkurrenz-Präparaten Gaboxadol, Indiplon und anderen. Dass der Markt aber weiterhin an diesem Indikationsgebiet Interesse zeigt, beweist der von Actelion im Juli verkündete Vertrag mit GSK zur gemeinsamen Entwicklung ihres Schlafmittels Almorexant. Erfolg in der Auslizenzierung ist für uns von enormer Bedeutung, um aus unseren Forschungsprojekten Wertschöpfung zu erzielen."
http://www.cash.ch/news/story/448/586933/40/40
Biotechfirma Evotec sucht nach finanzkräftigem Partner
19.08 09:08
HAMBURG (AWP International) - Das Biotech-Unternehmen Evotec sucht für die Entwicklung von Arzneien einen finanzkräftigen Partner aus der Pharmaindustrie. So will die Gesellschaft bis Jahresende für das am weitesten entwickelte Medikament, ein potenzielles Schlafmittel, einen "schlagkräftigen" Pharmakonzern als Partner gewonnen haben, berichtet die "Süddeutsche Zeitung" (SZ/Dienstagausgabe) unter Berufung auf Evotec-Chef Jörn Aldag. Das sei das wichtigste Jahresziel, denn Evotec plane 2009 den Start der dritten vorgeschriebenen Testphase. Das Investmenthaus Sal. Oppenheim traut dem Schlafmittel nach Zulassung Jahreserlöse in Höhe von bis zu 890 Millionen Euro zu.
Zudem forsche Evotec an einem Mittel zur Raucherentwöhnung, hiess es weiter. Dieses stecke seit Februar in Testphase II. Die Studie soll den Einfluss des Präparats auf das Suchtverhalten und die Entzugserscheinungen an 90 Rauchern zeigen. Für weitere kostspielige Tests sind die Hamburger ebenfalls auf Partner-Suche aus der Industrie. Insgesamt plant die Firma, dieses Jahr zwischen 46 und 51 Millionen Euro in die Forschung zu stecken. Die diesjährigen Umsätze dürften bei 30 bis 35 Millionen Euro liegen, sagte Aldag der Zeitung.
mfg ipollit
Antwort auf Beitrag Nr.: 34.759.628 von ipollit am 19.08.08 10:12:2206.08.2008 07:53
Hugin-News: Evotec AG
Evotec berichtet Geschäftszahlen für das erste Halbjahr 2008
Hamburg, Deutschland - Evotec AG (News/Aktienkurs) (Deutsche Börse: EVT; NASDAQ: EVTC) gab heute die Finanzergebnisse und den Verlauf des operativen Geschäfts im ersten Halbjahr 2008 bekannt.
Evotec erzielte im ersten Halbjahr 2008 Umsatzerlöse in Höhe von 14,5 Mio. Euro. Dies entspricht einem Rückgang um 8% gegenüber dem Vorjahr (2007: 15,8 Mio. Euro). Den Ausschlag für diese Entwicklung gaben in erster Linie Wechselkurseffekte, und zwar vor allem die weitere Schwächung des US-Dollars gegenüber Evotecs Berichtswährung, dem Euro. Bei gegenüber 2007 unveränderten Wechselkursen (britisches Pfund und US-Dollar) hätte der Umsatz im ersten Halbjahr 2008 16,1 Mio. Euro betragen und damit leicht über dem des ersten Halbjahrs 2007 gelegen.
Die hohen Investitionen in Forschung und Entwicklung führten zu einem höheren operativen Verlust von 26,9 Mio. Euro (erstes Halbjahr 2007: 23,6 Mio. Euro). Insgesamt stiegen die Forschungs- und Entwicklungsaufwendungen um etwa ein Drittel (33%) auf 21,9 Mio. Euro (erstes Halbjahr 2007: 16,4 Mio. Euro). Für diese Entwicklung waren vor allem zwei Faktoren verantwortlich: Die im ersten Quartal 2008 verbuchte Meilensteinzahlung an Roche für den Beginn der klinischen Phase-II-Studien mit EVT 302 sowie die Einbeziehung von Renovis nach der Akquisition im Mai.
Der Bestand an liquiden Mitteln hat sich nach der Akquisition von Renovis auf 101,0 Mio. Euro per 30. Juni 2008 erhöht (31. Dezember 2007: 93,7 Mio. Euro). Er umfasst Bargeld (42,4 Mio. Euro), kurzfristige Wertpapiere (50,3 Mio. Euro) sowie "Auction Rate Securities" (8,3 Mio. Euro). Auf Basis der Wechselkurse vom Jahresende 2007 hätte die Liquidität per 30. Juni 2008 105,6 Mio. Euro betragen. Evotec einschließlich Renovis erfuhr einen nicht realisierten Bilanzverlust von etwa 4,6 Mio. Euro durch die Umrechnung von in US-Dollar oder in britischen Pfund gehaltenen liquiden Mitteln in Euro.
Renovis-Akquisition erfolgreich abgeschlossen Am 1. Mai 2008 stimmten die Renovis-Aktionäre zugunsten der Übernahme durch Evotec. Diese wurde daraufhin am 2. Mai 2008 abgeschlossen. Die Aktionäre von Renovis erhielten für jede ausstehende Stammaktie von Renovis 0,5271 American Depositary Shares, kurz ADSs, von Evotec. Die Evotec-ADSs wurden für den Handel am NASDAQ Global Market unter dem Börsenkürzel "EVTC" zugelassen. Um diese Transaktion zu realisieren, gab Evotec insgesamt 34.970.268 neue Aktien aus, die den ADSs, welche an die Renovis-Anteilseigner ausgegeben wurden, zugrunde liegen. Um sicherzustellen, dass die neuen Evotec-Aktionäre an der diesjährigen Hauptversammlung teilnehmen können, hat Evotec die Versammlung auf den 28. August 2008 verschoben.
Fortschritte in der klinischen Entwicklung
* Am 3. Juli 2008 gab Evotec die Ergebnisse zu den wesentlichen Endpunkten einer doppelt verblindeten, vierwöchigen Phase-Ib-Studie mit EVT 101 bekannt. EVT 101 ist ein oral verfügbarer, für den NR2B-Subtyp des NMDA-Rezeptors selektiver Antagonist mit Potenzial für die Behandlung der Alzheimer'schen Erkrankung, neuropathischer Schmerzen sowie anderer Erkrankungen. Die Studie hat gezeigt, dass der Wirkstoff sowohl von jüngeren als auch von älteren Probanden bis zur höchsten getesteten Dosierung gut vertragen wurde.
* Am 1. August 2008 meldete Evotec, dass Pfizer Inc. im Rahmen ihrer Kooperation mit Evotec eine klinische Phase-I-Studie mit einem niedermolekularen VR1-(Vanilliod-Rezeptor-1)-Antagonisten begonnen hat. In dieser Phase-I-Studie wird der Wirkstoff an gesunde Probanden verabreicht, um die Sicherheit, Verträglichkeit und das pharmakokinetische Profil der Substanz nach oraler Verabreichung zu untersuchen.
* Evotec wird die Ergebnisse der ersten Phase-II-Studie mit EVT 302, einem hoch selektiven und reversiblen MAO-B-Inhibitor zur Unterstützung der Raucherentwöhnung, im dritten Quartal 2008 bekannt geben. Für die relevante Wirksamkeitsstudie, die untersucht, in welchem Ausmaß starke Raucher unter dem Einfluss von EVT 302 das Rauchen aufgeben, haben wir die Genehmigung der Ethikkommission erhalten und erwarten die endgültige Zulassung in Kürze.
* Evotecs proprietärer P2X7-Antagonist ist auf einem guten Weg, um im dritten Quartal 2008 in die Phase I zu gelangen. Wir werden in Kürze den Zulassungsantrag stellen, um die erste Studie im Menschen durchzuführen, in der Sicherheit, Verträglichkeit, pharmakokinetische und pharmakodynamische Eigenschaften unseres oral verfügbaren P2X7-Antagonisten untersucht werden.
Jörn Aldag, Vorstandsvorsitzender von Evotec, kommentierte dazu: "Im zweiten Quartal sind unsere klinischen Entwicklungsprogramme weiter gut vorangeschritten. Darüber hinaus sind wir sehr erfreut, dass Pfizer eine Phase-I-Studie mit unserem VR1-Rezeptor-Antagonisten, einem Programm aus der Akquisition von Renovis, begonnen hat. Mit dieser Akquisition haben wir einen wichtigen Meilenstein in unserer strategischen Entwicklung zu einem biopharmazeutischen Unternehmen erreicht. Unsere vier klinischen Wirkstoffkandidaten sind nun durch eine starke Pipeline im späten präklinischen Stadium untermauert, und wir sind heute gut positioniert, um für unsere Hauptprogramme Pharmakonzerne als Partner zu gewinnen. Entsprechende Gespräche für unser Produkt EVT 201 zur Behandlung von Schlafstörungen laufen. Schlaflosigkeit bleibt hinsichtlich des Abschlusses neuer Kooperationen ein schwieriges Indikationsgebiet, insbesondere nach den jüngsten Rückschlägen bei den Konkurrenz-Präparaten Gaboxadol, Indiplon und anderen. Dass der Markt aber weiterhin an diesem Indikationsgebiet Interesse zeigt, beweist der von Actelion im Juli verkündete Vertrag mit GSK zur gemeinsamen Entwicklung ihres Schlafmittels Almorexant. Erfolg in der Auslizenzierung ist für uns von enormer Bedeutung, um aus unseren Forschungsprojekten Wertschöpfung zu erzielen."
Finanzprognose 2008 Wir erwarten für das Geschäftsjahr 2008 Umsätze in der Größenordnung von 34 bis 36 Mio. Euro. Erlöse aus einem möglichen Auslizenzierungsabkommen sind darin noch nicht berücksichtigt. Das operative Ergebnis ohne Berücksichtigung von Erlösen durch Auslizenzierungen und ohne Wertberichtigungen im Jahr 2008 soll ungefähr auf dem Niveau von 2007 liegen. Im Fall einer erfolgreichen Auslizenzierung könnte es das Geschäftsergebnis von 2007 deutlich übertreffen. Evotec investiert weiterhin in Forschung und Entwicklung (F+E). Das Unternehmen geht davon aus, dass die F+E-Aufwendungen im Geschäftsjahr 2008 das untere Ende der Prognose von 46 bis 51 Mio. Euro erreichen werden. Hinzu kommen Aufwendungen für Aktienoptionsprogramme für Mitarbeiter, die den F+E-Aufwendungen 2008 zugeordnet werden. Der Anstieg gegenüber 2007 ist überwiegend auf den Fortschritt der klinischen Pipeline sowie die Einbeziehung von Renovis zurückzuführen. Unsere Liquiditätsprognose besagte, dass unsere liquiden Mittel zum Jahresende 2008 auf Basis konstanter Wechselkurse gegenüber Ende 2007 über 85 Mio. Euro betragen werden. Darin waren keine positiven Beiträge aus potenziellen Erlösen aus Auslizenzierungen enthalten. Diese Jahresendprognose beläuft sich unter Anwendung aktueller Wechselkurse heute auf knapp über 80 Mio. Euro. Wie bereits oben erwähnt, erfasste Evotec einschließlich Renovis in der ersten Jahreshälfte 2008 einen nicht realisierten Bilanzverlust von etwa 4,6 Mio. Euro durch die Umrechnung von in US-Dollar oder in britischen Pfund gehaltenen liquiden Mitteln in Euro. Unsere Liquiditätsprognose auf Basis aktueller Wechselkurse beträgt daher 80 Mio. Euro und kann trotz der Zahlung von 2,7 Mio. Euro an Roche, die wir im Juli 2008 für den Start der Phase II mit EVT 302 geleistet haben, aufrecht erhalten werden. Wir begrüßen die Zahlung an Roche in bar statt in Aktien, da dies den Anteilsbesitz unserer Aktionäre auf dem aktuell niedrigen Kursniveau nicht weiter verwässert. Unter der Annahme, dass sich das Portfolio des Unternehmens wie geplant weiterentwickelt und selbst wenn keine wichtigen Auslizenzierungen abgeschlossen werden, sollten die liquiden Mittel ausreichen, um Evotecs Entwicklungsprogramme bis Ende 2010 zu finanzieren.
Alle in dieser Pressemitteilung ausgewiesenen Ergebnisse und Erläuterungen für das Jahr 2008 werden mit den fortgeführten Geschäftsbereichen im Jahr 2007 verglichen. Am 30. November 2007 hat Evotec einen größeren Geschäftsbereich, die chemische Entwicklungssparte, an die US-Firma Aptuit verkauft. Ab dem 1. Dezember 2007 wurde dieses Geschäft nicht mehr im Abschluss der Evotec-Gruppe konsolidiert. Erträge und Aufwendungen für dieses Geschäft wurden in der Gewinn- und Verlust-Rechnung rückwirkend als aufzugebende Geschäftsbereiche ausgewiesen. Darüber hinaus hat Evotec am 2. Mai 2008 die Akquisition von Renovis, Inc. abgeschlossen. Die operativen Ergebnisse von Renovis sind daher für den Zeitraum vom 2. Mai 2008 bis 30. Juni 2008 in den konsolidierten Zwischen-Gewinn- und Verlustrechnungen für die ersten sechs Monate 2008 berücksichtigt sowie das Vermögen und die Verbindlichkeiten von Renovis per 30. Juni 2008 in der konsolidierten Zwischenbilanz enthalten. Daher sind die Ergebnisse der Jahre 2007 und 2008 nicht vollständig vergleichbar.
mfg ipollit
Hugin-News: Evotec AG
Evotec berichtet Geschäftszahlen für das erste Halbjahr 2008
Hamburg, Deutschland - Evotec AG (News/Aktienkurs) (Deutsche Börse: EVT; NASDAQ: EVTC) gab heute die Finanzergebnisse und den Verlauf des operativen Geschäfts im ersten Halbjahr 2008 bekannt.
Evotec erzielte im ersten Halbjahr 2008 Umsatzerlöse in Höhe von 14,5 Mio. Euro. Dies entspricht einem Rückgang um 8% gegenüber dem Vorjahr (2007: 15,8 Mio. Euro). Den Ausschlag für diese Entwicklung gaben in erster Linie Wechselkurseffekte, und zwar vor allem die weitere Schwächung des US-Dollars gegenüber Evotecs Berichtswährung, dem Euro. Bei gegenüber 2007 unveränderten Wechselkursen (britisches Pfund und US-Dollar) hätte der Umsatz im ersten Halbjahr 2008 16,1 Mio. Euro betragen und damit leicht über dem des ersten Halbjahrs 2007 gelegen.
Die hohen Investitionen in Forschung und Entwicklung führten zu einem höheren operativen Verlust von 26,9 Mio. Euro (erstes Halbjahr 2007: 23,6 Mio. Euro). Insgesamt stiegen die Forschungs- und Entwicklungsaufwendungen um etwa ein Drittel (33%) auf 21,9 Mio. Euro (erstes Halbjahr 2007: 16,4 Mio. Euro). Für diese Entwicklung waren vor allem zwei Faktoren verantwortlich: Die im ersten Quartal 2008 verbuchte Meilensteinzahlung an Roche für den Beginn der klinischen Phase-II-Studien mit EVT 302 sowie die Einbeziehung von Renovis nach der Akquisition im Mai.
Der Bestand an liquiden Mitteln hat sich nach der Akquisition von Renovis auf 101,0 Mio. Euro per 30. Juni 2008 erhöht (31. Dezember 2007: 93,7 Mio. Euro). Er umfasst Bargeld (42,4 Mio. Euro), kurzfristige Wertpapiere (50,3 Mio. Euro) sowie "Auction Rate Securities" (8,3 Mio. Euro). Auf Basis der Wechselkurse vom Jahresende 2007 hätte die Liquidität per 30. Juni 2008 105,6 Mio. Euro betragen. Evotec einschließlich Renovis erfuhr einen nicht realisierten Bilanzverlust von etwa 4,6 Mio. Euro durch die Umrechnung von in US-Dollar oder in britischen Pfund gehaltenen liquiden Mitteln in Euro.
Renovis-Akquisition erfolgreich abgeschlossen Am 1. Mai 2008 stimmten die Renovis-Aktionäre zugunsten der Übernahme durch Evotec. Diese wurde daraufhin am 2. Mai 2008 abgeschlossen. Die Aktionäre von Renovis erhielten für jede ausstehende Stammaktie von Renovis 0,5271 American Depositary Shares, kurz ADSs, von Evotec. Die Evotec-ADSs wurden für den Handel am NASDAQ Global Market unter dem Börsenkürzel "EVTC" zugelassen. Um diese Transaktion zu realisieren, gab Evotec insgesamt 34.970.268 neue Aktien aus, die den ADSs, welche an die Renovis-Anteilseigner ausgegeben wurden, zugrunde liegen. Um sicherzustellen, dass die neuen Evotec-Aktionäre an der diesjährigen Hauptversammlung teilnehmen können, hat Evotec die Versammlung auf den 28. August 2008 verschoben.
Fortschritte in der klinischen Entwicklung
* Am 3. Juli 2008 gab Evotec die Ergebnisse zu den wesentlichen Endpunkten einer doppelt verblindeten, vierwöchigen Phase-Ib-Studie mit EVT 101 bekannt. EVT 101 ist ein oral verfügbarer, für den NR2B-Subtyp des NMDA-Rezeptors selektiver Antagonist mit Potenzial für die Behandlung der Alzheimer'schen Erkrankung, neuropathischer Schmerzen sowie anderer Erkrankungen. Die Studie hat gezeigt, dass der Wirkstoff sowohl von jüngeren als auch von älteren Probanden bis zur höchsten getesteten Dosierung gut vertragen wurde.
* Am 1. August 2008 meldete Evotec, dass Pfizer Inc. im Rahmen ihrer Kooperation mit Evotec eine klinische Phase-I-Studie mit einem niedermolekularen VR1-(Vanilliod-Rezeptor-1)-Antagonisten begonnen hat. In dieser Phase-I-Studie wird der Wirkstoff an gesunde Probanden verabreicht, um die Sicherheit, Verträglichkeit und das pharmakokinetische Profil der Substanz nach oraler Verabreichung zu untersuchen.
* Evotec wird die Ergebnisse der ersten Phase-II-Studie mit EVT 302, einem hoch selektiven und reversiblen MAO-B-Inhibitor zur Unterstützung der Raucherentwöhnung, im dritten Quartal 2008 bekannt geben. Für die relevante Wirksamkeitsstudie, die untersucht, in welchem Ausmaß starke Raucher unter dem Einfluss von EVT 302 das Rauchen aufgeben, haben wir die Genehmigung der Ethikkommission erhalten und erwarten die endgültige Zulassung in Kürze.
* Evotecs proprietärer P2X7-Antagonist ist auf einem guten Weg, um im dritten Quartal 2008 in die Phase I zu gelangen. Wir werden in Kürze den Zulassungsantrag stellen, um die erste Studie im Menschen durchzuführen, in der Sicherheit, Verträglichkeit, pharmakokinetische und pharmakodynamische Eigenschaften unseres oral verfügbaren P2X7-Antagonisten untersucht werden.
Jörn Aldag, Vorstandsvorsitzender von Evotec, kommentierte dazu: "Im zweiten Quartal sind unsere klinischen Entwicklungsprogramme weiter gut vorangeschritten. Darüber hinaus sind wir sehr erfreut, dass Pfizer eine Phase-I-Studie mit unserem VR1-Rezeptor-Antagonisten, einem Programm aus der Akquisition von Renovis, begonnen hat. Mit dieser Akquisition haben wir einen wichtigen Meilenstein in unserer strategischen Entwicklung zu einem biopharmazeutischen Unternehmen erreicht. Unsere vier klinischen Wirkstoffkandidaten sind nun durch eine starke Pipeline im späten präklinischen Stadium untermauert, und wir sind heute gut positioniert, um für unsere Hauptprogramme Pharmakonzerne als Partner zu gewinnen. Entsprechende Gespräche für unser Produkt EVT 201 zur Behandlung von Schlafstörungen laufen. Schlaflosigkeit bleibt hinsichtlich des Abschlusses neuer Kooperationen ein schwieriges Indikationsgebiet, insbesondere nach den jüngsten Rückschlägen bei den Konkurrenz-Präparaten Gaboxadol, Indiplon und anderen. Dass der Markt aber weiterhin an diesem Indikationsgebiet Interesse zeigt, beweist der von Actelion im Juli verkündete Vertrag mit GSK zur gemeinsamen Entwicklung ihres Schlafmittels Almorexant. Erfolg in der Auslizenzierung ist für uns von enormer Bedeutung, um aus unseren Forschungsprojekten Wertschöpfung zu erzielen."
Finanzprognose 2008 Wir erwarten für das Geschäftsjahr 2008 Umsätze in der Größenordnung von 34 bis 36 Mio. Euro. Erlöse aus einem möglichen Auslizenzierungsabkommen sind darin noch nicht berücksichtigt. Das operative Ergebnis ohne Berücksichtigung von Erlösen durch Auslizenzierungen und ohne Wertberichtigungen im Jahr 2008 soll ungefähr auf dem Niveau von 2007 liegen. Im Fall einer erfolgreichen Auslizenzierung könnte es das Geschäftsergebnis von 2007 deutlich übertreffen. Evotec investiert weiterhin in Forschung und Entwicklung (F+E). Das Unternehmen geht davon aus, dass die F+E-Aufwendungen im Geschäftsjahr 2008 das untere Ende der Prognose von 46 bis 51 Mio. Euro erreichen werden. Hinzu kommen Aufwendungen für Aktienoptionsprogramme für Mitarbeiter, die den F+E-Aufwendungen 2008 zugeordnet werden. Der Anstieg gegenüber 2007 ist überwiegend auf den Fortschritt der klinischen Pipeline sowie die Einbeziehung von Renovis zurückzuführen. Unsere Liquiditätsprognose besagte, dass unsere liquiden Mittel zum Jahresende 2008 auf Basis konstanter Wechselkurse gegenüber Ende 2007 über 85 Mio. Euro betragen werden. Darin waren keine positiven Beiträge aus potenziellen Erlösen aus Auslizenzierungen enthalten. Diese Jahresendprognose beläuft sich unter Anwendung aktueller Wechselkurse heute auf knapp über 80 Mio. Euro. Wie bereits oben erwähnt, erfasste Evotec einschließlich Renovis in der ersten Jahreshälfte 2008 einen nicht realisierten Bilanzverlust von etwa 4,6 Mio. Euro durch die Umrechnung von in US-Dollar oder in britischen Pfund gehaltenen liquiden Mitteln in Euro. Unsere Liquiditätsprognose auf Basis aktueller Wechselkurse beträgt daher 80 Mio. Euro und kann trotz der Zahlung von 2,7 Mio. Euro an Roche, die wir im Juli 2008 für den Start der Phase II mit EVT 302 geleistet haben, aufrecht erhalten werden. Wir begrüßen die Zahlung an Roche in bar statt in Aktien, da dies den Anteilsbesitz unserer Aktionäre auf dem aktuell niedrigen Kursniveau nicht weiter verwässert. Unter der Annahme, dass sich das Portfolio des Unternehmens wie geplant weiterentwickelt und selbst wenn keine wichtigen Auslizenzierungen abgeschlossen werden, sollten die liquiden Mittel ausreichen, um Evotecs Entwicklungsprogramme bis Ende 2010 zu finanzieren.
Alle in dieser Pressemitteilung ausgewiesenen Ergebnisse und Erläuterungen für das Jahr 2008 werden mit den fortgeführten Geschäftsbereichen im Jahr 2007 verglichen. Am 30. November 2007 hat Evotec einen größeren Geschäftsbereich, die chemische Entwicklungssparte, an die US-Firma Aptuit verkauft. Ab dem 1. Dezember 2007 wurde dieses Geschäft nicht mehr im Abschluss der Evotec-Gruppe konsolidiert. Erträge und Aufwendungen für dieses Geschäft wurden in der Gewinn- und Verlust-Rechnung rückwirkend als aufzugebende Geschäftsbereiche ausgewiesen. Darüber hinaus hat Evotec am 2. Mai 2008 die Akquisition von Renovis, Inc. abgeschlossen. Die operativen Ergebnisse von Renovis sind daher für den Zeitraum vom 2. Mai 2008 bis 30. Juni 2008 in den konsolidierten Zwischen-Gewinn- und Verlustrechnungen für die ersten sechs Monate 2008 berücksichtigt sowie das Vermögen und die Verbindlichkeiten von Renovis per 30. Juni 2008 in der konsolidierten Zwischenbilanz enthalten. Daher sind die Ergebnisse der Jahre 2007 und 2008 nicht vollständig vergleichbar.
mfg ipollit
Antwort auf Beitrag Nr.: 34.759.628 von ipollit am 19.08.08 10:12:22http://www.boerse-online.de/aktien/deutschland_europa/:Evote…
EVOTEC - Unterbewerteter Biotechtitel
[14:15, 06.08.08]
Von Erich Gerbl
Der Hamburger Biotechkonzern Evotec sitzt auf einer vielversprechenden Produktpipeline. Die Bewertung an der Börse liegt dennoch nicht allzu fern vom Cashbestand. Die Aktie ist damit deutlich unterbewertet und eine Kaufgelegenheit.
Dem Hamburger Biotechunternehmen Evotec haben steigende Investitionen in die Medikamentenforschug im ersten Halbjahr einen Netto-Verlust von 25,9 Millionen Euro beschert. Im Vergleich zur entsprechenden Vorjahresperiode ist das ein Zuwachs von 17 Prozent. Wegen des starken Euros ging der Umsatz auf der anderen Seite um acht Prozent auf 14,5 Millionen Euro zurück.
Die wesentliche Kennzahl bei Biotech-Unternehmen ist der Cashbestand. Ende Juni hatte Evotec noch 101 Millionen Euro an liquiden Mittel zur Verfügung. Die Mittel umfassen Bargeld (42,4 Millionen Euro), kurzfristige Wertpapiere (50,3 Millionen Euro) sowie "Auction Rate Securities" (8,3 Millionen Euro). „Selbst wenn wir keinen Partner für einen unserer Produktkandidaten finden, reicht das Geld bis zum 31. Dezember 2010. Aber von diesem Szenario gehe ich auf keinen Fall aus“, sagt Evotec-Chef Jörn Aldag gegenüber BÖRSE ONLINE. An der Börse wird Evotec derzeit mit knapp 130 Millionen Euro bewertet – im Vergleich zum Cashbestand eine vertretbare Relation.
Produktpipeline mit Zukauf vergrößert
Dabei hat sich die Zahl der Produktkandidaten mit der Übernahme des auf Schmerzmedikation spezialisierten US-Unternehmens Renovis noch deutlich vergrößert. Für Renovis wurde lediglich der Cashbestand bezahlt. Der Grund lag im größten Hoffnungsträger der Firma, einem Produkt gegen Schlaganfall, das gefloppt ist. Dennoch war der Zukauf eine gute Entscheidung, denn Evotec kann auf die 65 Mitarbeiter und drei verbliebenen Produktkandidaten von Renovis zurückgreifen. Ein Schmerzmittel von Renovis ist mit dem Partner Pfizer vor wenigen Tagen in die erste Phase der klinischen Tests gestartet. Damit hat Evotec zwei Kandidaten in der ersten und zwei Kandidaten in der zweiten klinischen Phase.
Schlafmittel mit Potenzial zum Blockbuster
Am weitesten fortgeschritten ist das Schlafmittel EVT 201. Anders als bei Konkurrenzprodukten hilft das Mittel EVT 201 sowohl beim Einschlafen als auch beim Durchschlafen. Noch dazu ist es gut verträglich. Die Wirksamkeit konnte in Phase II bereits bewiesen werden. Danach soll hier ein Marktpotential von 500 Millionen bis 1,1 Milliarden Euro schlummern. „Wir haben ein Produkt mit einem überlegenen Sicherheits- und Wirksamkeitsprofil" sagt Aldag. Dennoch sei der Schlafmittelmarkt kein einfacher. Generika drücken in diesem Bereich die Verkaufspreise.
Zudem sind zur Produkteinführung enorm hohe Marketingkosten nötig, um das Produkt und dessen Vorteile zu kommunizieren. Evotec-Chef Aldag erwartet sich eine zweistellige Umsatzbeteiligung. „Bis zum Jahresende wollen wir einen Partner finden“, so Aldag weiter. Auf den Markt soll das Produkt 2012 oder 2013 kommen. Mit Meilensteinzahlungen des neuen Partners wäre Evotec bis dahin jedoch finanziell abgesichert. Ein Unsicherheitsfaktor bringt das Mittel dennoch mit sich: wirtschaftlich könnte es ein nicht ganz so hohes Potential haben, da nur bestimmte Bevölkerungsgruppen auf das Produkt ansprechen.
Hinter EVT 201 rangiert EVT 302. Das ist ein potentielles Medikament zur Raucherentwöhnung. Während die auf dem Markt bereits verfügbaren Präparate auf den Botenstoff Dopamin setzen, blockt das Evotec-Produkt hingegen den Abbau von Dopamin ab. In Kürze startet eine zentrale Studie, die voraussichtlich im ersten Halbjahr 2009 erste Ergebnisse bringen wird.
Das Alzheimermedikament EVT 101 befindet sich in der ersten klinischen Phase. Zusätzlich werden die Renovis-Produkte P2X7 (Gelenkrheumatismus) und P2X3 (Schmerzen, Harninkontinenz) laut Aldag voraussichtlich im dritten Quartal 2008 und im zweiten Halbjahr 2009 in die klinische Phase gehen und den Wert von Evotec damit steigern. Während Produktkandidaten in der vorklinischen Phase von Investoren kein Wert zugemessen wird, werden Medikamente in der klinischen Phase bewertet.
BÖRSE ONLINE empfiehlt die Evotec-Aktie zum KAUF. Anders als die Börsenkapitalisierung das nahe legt, haben die Produkte eine reale Chance auf eine Markteinführung. Das Unternehmen ist damit klar unterbewertet. Obwohl der Fokus auf EVT 201 liegt, ist das Unternehmen mit vier Produkten in der klinischen Phase breit aufgestellt. Da die Wirksamkeit bereits bewiesen wurde, sind die Chancen, dass Evotec für EVT 201 einen Partner findet gut.
mfg ipollit
EVOTEC - Unterbewerteter Biotechtitel
[14:15, 06.08.08]
Von Erich Gerbl
Der Hamburger Biotechkonzern Evotec sitzt auf einer vielversprechenden Produktpipeline. Die Bewertung an der Börse liegt dennoch nicht allzu fern vom Cashbestand. Die Aktie ist damit deutlich unterbewertet und eine Kaufgelegenheit.
Dem Hamburger Biotechunternehmen Evotec haben steigende Investitionen in die Medikamentenforschug im ersten Halbjahr einen Netto-Verlust von 25,9 Millionen Euro beschert. Im Vergleich zur entsprechenden Vorjahresperiode ist das ein Zuwachs von 17 Prozent. Wegen des starken Euros ging der Umsatz auf der anderen Seite um acht Prozent auf 14,5 Millionen Euro zurück.
Die wesentliche Kennzahl bei Biotech-Unternehmen ist der Cashbestand. Ende Juni hatte Evotec noch 101 Millionen Euro an liquiden Mittel zur Verfügung. Die Mittel umfassen Bargeld (42,4 Millionen Euro), kurzfristige Wertpapiere (50,3 Millionen Euro) sowie "Auction Rate Securities" (8,3 Millionen Euro). „Selbst wenn wir keinen Partner für einen unserer Produktkandidaten finden, reicht das Geld bis zum 31. Dezember 2010. Aber von diesem Szenario gehe ich auf keinen Fall aus“, sagt Evotec-Chef Jörn Aldag gegenüber BÖRSE ONLINE. An der Börse wird Evotec derzeit mit knapp 130 Millionen Euro bewertet – im Vergleich zum Cashbestand eine vertretbare Relation.
Produktpipeline mit Zukauf vergrößert
Dabei hat sich die Zahl der Produktkandidaten mit der Übernahme des auf Schmerzmedikation spezialisierten US-Unternehmens Renovis noch deutlich vergrößert. Für Renovis wurde lediglich der Cashbestand bezahlt. Der Grund lag im größten Hoffnungsträger der Firma, einem Produkt gegen Schlaganfall, das gefloppt ist. Dennoch war der Zukauf eine gute Entscheidung, denn Evotec kann auf die 65 Mitarbeiter und drei verbliebenen Produktkandidaten von Renovis zurückgreifen. Ein Schmerzmittel von Renovis ist mit dem Partner Pfizer vor wenigen Tagen in die erste Phase der klinischen Tests gestartet. Damit hat Evotec zwei Kandidaten in der ersten und zwei Kandidaten in der zweiten klinischen Phase.
Schlafmittel mit Potenzial zum Blockbuster
Am weitesten fortgeschritten ist das Schlafmittel EVT 201. Anders als bei Konkurrenzprodukten hilft das Mittel EVT 201 sowohl beim Einschlafen als auch beim Durchschlafen. Noch dazu ist es gut verträglich. Die Wirksamkeit konnte in Phase II bereits bewiesen werden. Danach soll hier ein Marktpotential von 500 Millionen bis 1,1 Milliarden Euro schlummern. „Wir haben ein Produkt mit einem überlegenen Sicherheits- und Wirksamkeitsprofil" sagt Aldag. Dennoch sei der Schlafmittelmarkt kein einfacher. Generika drücken in diesem Bereich die Verkaufspreise.
Zudem sind zur Produkteinführung enorm hohe Marketingkosten nötig, um das Produkt und dessen Vorteile zu kommunizieren. Evotec-Chef Aldag erwartet sich eine zweistellige Umsatzbeteiligung. „Bis zum Jahresende wollen wir einen Partner finden“, so Aldag weiter. Auf den Markt soll das Produkt 2012 oder 2013 kommen. Mit Meilensteinzahlungen des neuen Partners wäre Evotec bis dahin jedoch finanziell abgesichert. Ein Unsicherheitsfaktor bringt das Mittel dennoch mit sich: wirtschaftlich könnte es ein nicht ganz so hohes Potential haben, da nur bestimmte Bevölkerungsgruppen auf das Produkt ansprechen.
Hinter EVT 201 rangiert EVT 302. Das ist ein potentielles Medikament zur Raucherentwöhnung. Während die auf dem Markt bereits verfügbaren Präparate auf den Botenstoff Dopamin setzen, blockt das Evotec-Produkt hingegen den Abbau von Dopamin ab. In Kürze startet eine zentrale Studie, die voraussichtlich im ersten Halbjahr 2009 erste Ergebnisse bringen wird.
Das Alzheimermedikament EVT 101 befindet sich in der ersten klinischen Phase. Zusätzlich werden die Renovis-Produkte P2X7 (Gelenkrheumatismus) und P2X3 (Schmerzen, Harninkontinenz) laut Aldag voraussichtlich im dritten Quartal 2008 und im zweiten Halbjahr 2009 in die klinische Phase gehen und den Wert von Evotec damit steigern. Während Produktkandidaten in der vorklinischen Phase von Investoren kein Wert zugemessen wird, werden Medikamente in der klinischen Phase bewertet.
BÖRSE ONLINE empfiehlt die Evotec-Aktie zum KAUF. Anders als die Börsenkapitalisierung das nahe legt, haben die Produkte eine reale Chance auf eine Markteinführung. Das Unternehmen ist damit klar unterbewertet. Obwohl der Fokus auf EVT 201 liegt, ist das Unternehmen mit vier Produkten in der klinischen Phase breit aufgestellt. Da die Wirksamkeit bereits bewiesen wurde, sind die Chancen, dass Evotec für EVT 201 einen Partner findet gut.
mfg ipollit
Regeneron... VEGF Trap-Eye weitere PII-Daten
Ziel von VEGF Trap-Eye ist nicht in erster Linie eine Verbesserung der Effektivität gegenüber des Lucentis/Avastin-AKs von Genentech, sondern die Verlängerung der Wirkung durch die gegenüber normalen AKs stabileren Traps. Dadurch müssen Patienten u.U. nicht mehr jeden Monat durch das Auge hindurch eine Dosis injiziert bekommen sondern z.B. nur alle paar Monate. Hier wurde z.B. nach den ersten festen Dosen im Schnitt nur alle ca. 6 Monate bzw. 4 Monate ins Auge gestochen.
************
19.08.2008 13:05
Regeneron and Bayer HealthCare Announce VEGF Trap-Eye Achieved Durable Improvement in Vision over 52 Weeks in a Phase 2 Study in Patients with Age-Related Macular Degeneration
Regeneron Pharmaceuticals, (News) Inc. (Nasdaq: REGN) and Bayer (News/Aktienkurs) HealthCare AG today announced that patients with wet age-related macular degeneration (AMD) receiving VEGF Trap-Eye in a Phase 2 extension study on a PRN (as needed) dosing schedule continued to show highly significant improvements at 52 weeks in the primary and key secondary endpoints of retinal thickness (an anatomic measure of treatment effect) and vision gain. The 12-week primary endpoint results from the fixed-dosing period of the study were presented at the 2007 Retina Society conference in September 2007. The 32-week results of the Phase 2 study were presented at the 2008 Association for Research in Vision and Ophthalmology (ARVO) meeting in Fort Lauderdale, Florida in April 2008. A full analysis of the 52-week results of the Phase 2 study will be presented at the 2008 meeting of the Retina Society on September 26-28, 2008 in Scottsdale, Arizona.
In this double-masked, prospective, randomized, multi-center Phase 2 trial, 157 patients were randomized to five dose groups and treated with VEGF Trap-Eye in one eye. Two groups initially received monthly doses of 0.5 or 2.0 milligrams (mg) of VEGF Trap-Eye (at weeks 0, 4, 8, and 12) and three groups received quarterly doses of 0.5, 2.0, or 4.0 mg of VEGF Trap-Eye (at baseline and week 12). Following the initial 12-week fixed-dosing phase of the trial, patients continued to receive therapy at the same dose on a PRN dosing schedule based upon the physician assessment of the need for re-treatment in accordance with pre-specified criteria. Patients were monitored for safety, retinal thickness, and visual acuity. These data represent the final one-year analysis from the 52-week study.
Patients receiving four monthly doses of VEGF Trap-Eye, either 2.0 or 0.5 mg, for 12 weeks followed by PRN dosing thereafter, achieved mean improvements in visual acuity versus baseline of 9.0 letters (p<0.0001) and 5.4 letters (p=0.085), respectively, and mean decreases in retinal thickness versus baseline of 143 microns (p<0.0001) and 125 microns (p<0.0001) at week 52, respectively. During the subsequent PRN dosing phase, patients initially dosed on a 2.0 mg monthly schedule received, on average, only 1.6 additional injections and those initially dosed on a 0.5 mg monthly schedule received, on average, 2.5 injections.
For all dose cohorts combined, there was a 5.3 mean letter gain in visual acuity versus baseline at the week 52 evaluation visit (p<0.0001). The mean decrease in retinal thickness for all dose groups combined at week 52 was 130 microns versus baseline (p<0.0001). During the week 12 to week 52 PRN dosing period, patients from all dose groups combined received, on average, only two additional injections.
VEGF Trap-Eye was generally well tolerated and there were no drug-related serious adverse events. There was one reported case of culture-negative endophthalmitis/uveitis in the study eye and one arterial thrombotic event, neither of which was deemed to be drug-related. The most common adverse events were those typically associated with intravitreal injections.
"Based upon retinal physicians' feedback, there remains a significant unmet medical need for a treatment for wet AMD that can reliably improve visual acuity over time without the need for monthly intravitreal injections,“ said George D. Yancopoulos, M.D., Ph.D., President of Regeneron Research Laboratories. ”We are excited about these study findings and the potential for VEGF Trap-Eye to fulfill this need pending the results of our ongoing Phase 3 clinical studies.“
”The 52-week results underline that VEGF Trap-Eye has the potential to significantly reduce retinal thickness and improve vision,“ said Dr. Kemal Malik, member of the Bayer HealthCare Executive Committee responsible for product development. ”The further development of this compound is important for millions of people worldwide who suffer from this devastating ocular disease.“
mfg ipollit
Ziel von VEGF Trap-Eye ist nicht in erster Linie eine Verbesserung der Effektivität gegenüber des Lucentis/Avastin-AKs von Genentech, sondern die Verlängerung der Wirkung durch die gegenüber normalen AKs stabileren Traps. Dadurch müssen Patienten u.U. nicht mehr jeden Monat durch das Auge hindurch eine Dosis injiziert bekommen sondern z.B. nur alle paar Monate. Hier wurde z.B. nach den ersten festen Dosen im Schnitt nur alle ca. 6 Monate bzw. 4 Monate ins Auge gestochen.
************
19.08.2008 13:05
Regeneron and Bayer HealthCare Announce VEGF Trap-Eye Achieved Durable Improvement in Vision over 52 Weeks in a Phase 2 Study in Patients with Age-Related Macular Degeneration
Regeneron Pharmaceuticals, (News) Inc. (Nasdaq: REGN) and Bayer (News/Aktienkurs) HealthCare AG today announced that patients with wet age-related macular degeneration (AMD) receiving VEGF Trap-Eye in a Phase 2 extension study on a PRN (as needed) dosing schedule continued to show highly significant improvements at 52 weeks in the primary and key secondary endpoints of retinal thickness (an anatomic measure of treatment effect) and vision gain. The 12-week primary endpoint results from the fixed-dosing period of the study were presented at the 2007 Retina Society conference in September 2007. The 32-week results of the Phase 2 study were presented at the 2008 Association for Research in Vision and Ophthalmology (ARVO) meeting in Fort Lauderdale, Florida in April 2008. A full analysis of the 52-week results of the Phase 2 study will be presented at the 2008 meeting of the Retina Society on September 26-28, 2008 in Scottsdale, Arizona.
In this double-masked, prospective, randomized, multi-center Phase 2 trial, 157 patients were randomized to five dose groups and treated with VEGF Trap-Eye in one eye. Two groups initially received monthly doses of 0.5 or 2.0 milligrams (mg) of VEGF Trap-Eye (at weeks 0, 4, 8, and 12) and three groups received quarterly doses of 0.5, 2.0, or 4.0 mg of VEGF Trap-Eye (at baseline and week 12). Following the initial 12-week fixed-dosing phase of the trial, patients continued to receive therapy at the same dose on a PRN dosing schedule based upon the physician assessment of the need for re-treatment in accordance with pre-specified criteria. Patients were monitored for safety, retinal thickness, and visual acuity. These data represent the final one-year analysis from the 52-week study.
Patients receiving four monthly doses of VEGF Trap-Eye, either 2.0 or 0.5 mg, for 12 weeks followed by PRN dosing thereafter, achieved mean improvements in visual acuity versus baseline of 9.0 letters (p<0.0001) and 5.4 letters (p=0.085), respectively, and mean decreases in retinal thickness versus baseline of 143 microns (p<0.0001) and 125 microns (p<0.0001) at week 52, respectively. During the subsequent PRN dosing phase, patients initially dosed on a 2.0 mg monthly schedule received, on average, only 1.6 additional injections and those initially dosed on a 0.5 mg monthly schedule received, on average, 2.5 injections.
For all dose cohorts combined, there was a 5.3 mean letter gain in visual acuity versus baseline at the week 52 evaluation visit (p<0.0001). The mean decrease in retinal thickness for all dose groups combined at week 52 was 130 microns versus baseline (p<0.0001). During the week 12 to week 52 PRN dosing period, patients from all dose groups combined received, on average, only two additional injections.
VEGF Trap-Eye was generally well tolerated and there were no drug-related serious adverse events. There was one reported case of culture-negative endophthalmitis/uveitis in the study eye and one arterial thrombotic event, neither of which was deemed to be drug-related. The most common adverse events were those typically associated with intravitreal injections.
"Based upon retinal physicians' feedback, there remains a significant unmet medical need for a treatment for wet AMD that can reliably improve visual acuity over time without the need for monthly intravitreal injections,“ said George D. Yancopoulos, M.D., Ph.D., President of Regeneron Research Laboratories. ”We are excited about these study findings and the potential for VEGF Trap-Eye to fulfill this need pending the results of our ongoing Phase 3 clinical studies.“
”The 52-week results underline that VEGF Trap-Eye has the potential to significantly reduce retinal thickness and improve vision,“ said Dr. Kemal Malik, member of the Bayer HealthCare Executive Committee responsible for product development. ”The further development of this compound is important for millions of people worldwide who suffer from this devastating ocular disease.“
mfg ipollit
Antwort auf Beitrag Nr.: 34.755.539 von ipollit am 19.08.08 00:14:19hier noch etwas zu Viropharma's Cinryze und den HAE Konkurrenten...
Companies line up for hereditary angioedema market
NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 4 APRIL 2008
Brady Huggett
At the end of this month, the first therapeutic for hereditary angioedema (HAE) could receive approval for marketing in the US. By end of next year, there could be as many as five biotech products on the HAE market from separate companies. Being first to receive approval in a small, niche indication will ensure a competitive edge, but differences in mode of action, delivery and eligibility for orphan status for the different therapies, as well as the heterogeneity of the patient population itself, might mean later entrants could still be successful as the market becomes segmented.
Berlin-based Jerini is poised first in line to receive a decision from the US Food and Drug Administration (FDA). The company's Icatibant, a synthetic peptidomimetic drug licensed from Paris-headquartered Sanofi Aventis, has a Prescription Drug User Fee Act date of April 26. Approval is by no means certain, however, as the HAE field has a history of late-stage regulatory setbacks (Box 1).
HAE is caused by low serum levels of C1 esterase inhibitor (C1-INH). As well as inhibiting components of the fibrinolytic, clotting and kinin pathways, C1-INH also blocks the activation of C1 and the rest of the classic complement pathway by binding to C1r and C1s (sub-components of C1). Low levels of C1-INH lead to unchecked complement activation that is important in inflammation and the clearance of pathogens from the body. There are two types of HAE patients: most are type 1 and have levels of C1-INH <35% lower than normal; those with type 2 have normal or elevated levels of C1-INH, but the protein is nonfunctional. Either way, both types suffer attacks that swell the hands, feet, larynx, facial features and genitals, with gastro-intestinal swelling that sometimes causes vomiting, diarrhea and severe pain. The condition is rare but autosomal dominant.
One key problem for drug developers is how to estimate the HAE market size. A "pretty standard" estimated prevalence is 1 person with HAE in every 30,000 people, says Samir Singh, president of US operations at Pharming (headquartered in Leiden, The Netherlands). This suggests about 10,000 people in the US and 22,000 people in the Western world with HAE. "When you look at the US, Europe and Japan, that's total treatable attacks of 200,000 [annually]," he estimates.
All the players think the market is set to grow and that the disease is often mis- or underdiagnosed. Cambridge, Massachusetts–based Dyax's general counsel and executive vice president of administration, Ivana Magovevi-Liebisch, says diagnosing the disease can take as long as ten years, as patients repeatedly leave hospitals after the attack resolves without a physician fingering HAE as the culprit.
"Patients look like they are having an allergic reaction," she says, and even as they suffer vomiting and diarrhea, doctors "can't find anything wrong." Eventually an allergist suggests HAE, and the patient begins mining the family history to discover a pattern of illness in relatives.
Once diagnosed, there is no consensus on whether those individuals should receive acute or prophylactic care, mostly because the frequency of attacks is also unclear. CSL Behring of King of Prussia, Pennsylvania, already markets its C1-INH product Berinert in Germany, Austria and Switzerland, where the drug has been administered >300,000 times. Published data from these European patients point to about seven attacks per year, though many assume it's higher and Pharming's Singh says certain surveys have it pegged as high as 20 annually.
If people with HAE are having 20 or more attacks yearly, then "one could make a case for prophylactic use," says Singh, though personally he's doubtful. In fact, only New York-based Lev is developing its product, Cinryze, to include the prophylactic market. Dyax's executive vice president and chief business officer, Gustav Christensen, doubts that market exists. Prophylactic treatment could mean two injections a week, driving the cost per patient to $250,000 annually, so insurance carriers would probably begin pushing cheaper acute treatment as preferred.
Three of the five drugs vying for approval aim to address HAE by supplementing the individual's endogenous C1-INH with a functional version of the human protein (Table 1). Lev and CSL Behring are both developing a purified plasma protein version, whereas Pharming has developed a recombinant C1-INH produced in transgenic rabbits.
The other two companies—Dyax and Jerini—are developing proteins aimed at dampening the inflammatory cascade. Dyax's recombinant protein DX-88 (ecallantide) inhibits kallikrein, an enzyme that liberates bradykinin, which causes fluid to leak from blood vessels into the tissues. Jerini's Icatibant attacks one step further down the line; it's a competitive bradykinin B2 receptor antagonist. All five drugs are deemed comparably efficacious (though there have been no head-to-head clinical trials to help determine this), which means it's difficult to predict which product will have the competitive edge.
One differentiating factor is delivery. DX-88 is just 60 amino acids long and can be administered subcutaneously, as can Icatibant, but the C1-INH products must be given by intravenous (i.v.) infusion. Jerini CEO Jens Schneider-Mergener says that, based on feedback from physicians, his firm sees "a big advantage" in the subcutaneous route over the i.v. one. Bret Holley, analyst with New York-based Oppenheimer, which covers Lev but receives no compensation from the firm, tends to agree, venturing that i.v. infusion can be seen as a "killing handicap." But Holley also argues that attacks are foreseen by people with HAE, much like migraines, allowing them time to seek a physician for an infusion, and he points out that Dyax's DX-88 needs to be refrigerated anyway. So far, Jerini boasts the only subcutaneous administration that does not need to be refrigerated—the company has data showing sterility exceeding 18 months at room temperature. Dyax is working on a room-temperature formulation.
The final uncertainty is orphan drug status—all have it except CSL Behring. The FDA makes the final decision here, but it is assumed DX-88 and Icatibant will be viewed as independent molecules and thus each drug's orphan drug status will not block another from entering the market. Pharming's Samir says that his company's C1 inhibitor should stand alone, as it is a recombinant molecule. Whether the plasma-derived C1 inhibitors will be viewed as the same molecule isn't clear.
There is general consensus, however, on what's at risk: a $500 million-a-year market. The small patient population and unmet medical need means the community will tolerate a "Genzyme-like pricing," Holley says, referring to the empire Cambridge, Massachusetts-based Genzyme has built by developing niche products for rare diseases. The HAE market, Holley believes, will be shaped in much the same way as the one formed around Genzyme's products.
"Patients will be treated on a physician-by-physician basis," he says, so the first step is to "get a therapy out there." Only then, as patients choose the product that works best for them, physicians become comfortable and usage is bent to individual needs, will there be hints as to how these products will settle out.
mfg ipollit
Companies line up for hereditary angioedema market
NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 4 APRIL 2008
Brady Huggett
At the end of this month, the first therapeutic for hereditary angioedema (HAE) could receive approval for marketing in the US. By end of next year, there could be as many as five biotech products on the HAE market from separate companies. Being first to receive approval in a small, niche indication will ensure a competitive edge, but differences in mode of action, delivery and eligibility for orphan status for the different therapies, as well as the heterogeneity of the patient population itself, might mean later entrants could still be successful as the market becomes segmented.
Berlin-based Jerini is poised first in line to receive a decision from the US Food and Drug Administration (FDA). The company's Icatibant, a synthetic peptidomimetic drug licensed from Paris-headquartered Sanofi Aventis, has a Prescription Drug User Fee Act date of April 26. Approval is by no means certain, however, as the HAE field has a history of late-stage regulatory setbacks (Box 1).
HAE is caused by low serum levels of C1 esterase inhibitor (C1-INH). As well as inhibiting components of the fibrinolytic, clotting and kinin pathways, C1-INH also blocks the activation of C1 and the rest of the classic complement pathway by binding to C1r and C1s (sub-components of C1). Low levels of C1-INH lead to unchecked complement activation that is important in inflammation and the clearance of pathogens from the body. There are two types of HAE patients: most are type 1 and have levels of C1-INH <35% lower than normal; those with type 2 have normal or elevated levels of C1-INH, but the protein is nonfunctional. Either way, both types suffer attacks that swell the hands, feet, larynx, facial features and genitals, with gastro-intestinal swelling that sometimes causes vomiting, diarrhea and severe pain. The condition is rare but autosomal dominant.
One key problem for drug developers is how to estimate the HAE market size. A "pretty standard" estimated prevalence is 1 person with HAE in every 30,000 people, says Samir Singh, president of US operations at Pharming (headquartered in Leiden, The Netherlands). This suggests about 10,000 people in the US and 22,000 people in the Western world with HAE. "When you look at the US, Europe and Japan, that's total treatable attacks of 200,000 [annually]," he estimates.
All the players think the market is set to grow and that the disease is often mis- or underdiagnosed. Cambridge, Massachusetts–based Dyax's general counsel and executive vice president of administration, Ivana Magovevi-Liebisch, says diagnosing the disease can take as long as ten years, as patients repeatedly leave hospitals after the attack resolves without a physician fingering HAE as the culprit.
"Patients look like they are having an allergic reaction," she says, and even as they suffer vomiting and diarrhea, doctors "can't find anything wrong." Eventually an allergist suggests HAE, and the patient begins mining the family history to discover a pattern of illness in relatives.
Once diagnosed, there is no consensus on whether those individuals should receive acute or prophylactic care, mostly because the frequency of attacks is also unclear. CSL Behring of King of Prussia, Pennsylvania, already markets its C1-INH product Berinert in Germany, Austria and Switzerland, where the drug has been administered >300,000 times. Published data from these European patients point to about seven attacks per year, though many assume it's higher and Pharming's Singh says certain surveys have it pegged as high as 20 annually.
If people with HAE are having 20 or more attacks yearly, then "one could make a case for prophylactic use," says Singh, though personally he's doubtful. In fact, only New York-based Lev is developing its product, Cinryze, to include the prophylactic market. Dyax's executive vice president and chief business officer, Gustav Christensen, doubts that market exists. Prophylactic treatment could mean two injections a week, driving the cost per patient to $250,000 annually, so insurance carriers would probably begin pushing cheaper acute treatment as preferred.
Three of the five drugs vying for approval aim to address HAE by supplementing the individual's endogenous C1-INH with a functional version of the human protein (Table 1). Lev and CSL Behring are both developing a purified plasma protein version, whereas Pharming has developed a recombinant C1-INH produced in transgenic rabbits.
The other two companies—Dyax and Jerini—are developing proteins aimed at dampening the inflammatory cascade. Dyax's recombinant protein DX-88 (ecallantide) inhibits kallikrein, an enzyme that liberates bradykinin, which causes fluid to leak from blood vessels into the tissues. Jerini's Icatibant attacks one step further down the line; it's a competitive bradykinin B2 receptor antagonist. All five drugs are deemed comparably efficacious (though there have been no head-to-head clinical trials to help determine this), which means it's difficult to predict which product will have the competitive edge.
One differentiating factor is delivery. DX-88 is just 60 amino acids long and can be administered subcutaneously, as can Icatibant, but the C1-INH products must be given by intravenous (i.v.) infusion. Jerini CEO Jens Schneider-Mergener says that, based on feedback from physicians, his firm sees "a big advantage" in the subcutaneous route over the i.v. one. Bret Holley, analyst with New York-based Oppenheimer, which covers Lev but receives no compensation from the firm, tends to agree, venturing that i.v. infusion can be seen as a "killing handicap." But Holley also argues that attacks are foreseen by people with HAE, much like migraines, allowing them time to seek a physician for an infusion, and he points out that Dyax's DX-88 needs to be refrigerated anyway. So far, Jerini boasts the only subcutaneous administration that does not need to be refrigerated—the company has data showing sterility exceeding 18 months at room temperature. Dyax is working on a room-temperature formulation.
The final uncertainty is orphan drug status—all have it except CSL Behring. The FDA makes the final decision here, but it is assumed DX-88 and Icatibant will be viewed as independent molecules and thus each drug's orphan drug status will not block another from entering the market. Pharming's Samir says that his company's C1 inhibitor should stand alone, as it is a recombinant molecule. Whether the plasma-derived C1 inhibitors will be viewed as the same molecule isn't clear.
There is general consensus, however, on what's at risk: a $500 million-a-year market. The small patient population and unmet medical need means the community will tolerate a "Genzyme-like pricing," Holley says, referring to the empire Cambridge, Massachusetts-based Genzyme has built by developing niche products for rare diseases. The HAE market, Holley believes, will be shaped in much the same way as the one formed around Genzyme's products.
"Patients will be treated on a physician-by-physician basis," he says, so the first step is to "get a therapy out there." Only then, as patients choose the product that works best for them, physicians become comfortable and usage is bent to individual needs, will there be hints as to how these products will settle out.
mfg ipollit
Vertex - weitere PIII gestartet
ich bin mir nicht sicher, ob VRTX's Telaprevir deutlich vor Schering's Boceprevir auf den Markt kommen wird... außerdem scheint die Wirksamkeit usw. ähnlich zu sein. Aufgrund dieser Konkurrenz bin ich lieber etwas vorsichtig, auch wenn Telaprevir ein großer Erfolg werden kann (aber erst in 2 Jahren)... ich werde wohl meine Vertex-Position erstmal auflösen. (VRTX MK aktuell 4 Mrd USD, >800 Mio USD Cash, sehr hoher Cashburn)
http://www.thestreet.com/story/10433829/1/vertex-jj-to-begin…
Vertex, J&J to Begin New Telaprevir Study
08/19/08 - 08:33 AM EDT
Vertex Pharmaceuticals(VRTX - Cramer's Take - Stockpickr) announced Tuesday an agreement with regulators in the U.S. and Europe that greenlights the company and partner Johnson & Johnson(JNJ - Cramer's Take - Stockpickr) to begin a new phase III study of their experimental hepatitis C drug telaprevir.
The new study will test telaprevir with standard combination therapy in 650 hepatitis C patients who could not be cured with the currently approved drugs for the disease, pegylated interferon and ribavirin. These are difficult to treat patients, including patients known as "null responders," which means they did not respond at all to previous treatments.
Vertex and Johnson & Johnson, which are co-developing telaprevir, posted an outline of this new study, dubbed REALIZE, on the ClinicalTrials.gov Web site in June. Vertex shares fell at that time because the new phase III trial suggested that an early regulatory approval filing for telaprevir in 2009 for treatment-resistant patients was not in the cards.
As it stands now, Vertex and Johnson & Johnson won't likely seek approval for telaprevir until the second half of 2010, based on data from a separate phase III study of the drug in treatment-naïve hepatitis C patients.
Vertex and its partner have previously released results from a smaller study in treatment-resistant patients that showed telaprevir to be effective in attacking the virus that causes hepatitis C.
The new phase III study announced today will test two different regimens of telaprevir in patients who previously did not respond to conventional hepatitis C treatment with pegylated interferon and ribavirin.
The study differs from previous studies in that patients will be treated with two regimens of telaprevir for a full 48 weeks instead of the accelerated 24-week treatment cycle used for treatment-naïve patients.
In addition, one of the arms of the new study will see whether a "lead-in" treatment with interferon and ribavirin alone before dosing of telaprevir may improve overall cure rates.
Vertex and its partner are locked in a tight race with Schering-Plough(SGP - Cramer's Take - Stockpickr) to be the first with a new direct antiviral drug aimed at hepatitis C approved in the U.S. and Europe.
mfg ipollit
ich bin mir nicht sicher, ob VRTX's Telaprevir deutlich vor Schering's Boceprevir auf den Markt kommen wird... außerdem scheint die Wirksamkeit usw. ähnlich zu sein. Aufgrund dieser Konkurrenz bin ich lieber etwas vorsichtig, auch wenn Telaprevir ein großer Erfolg werden kann (aber erst in 2 Jahren)... ich werde wohl meine Vertex-Position erstmal auflösen. (VRTX MK aktuell 4 Mrd USD, >800 Mio USD Cash, sehr hoher Cashburn)
http://www.thestreet.com/story/10433829/1/vertex-jj-to-begin…
Vertex, J&J to Begin New Telaprevir Study
08/19/08 - 08:33 AM EDT
Vertex Pharmaceuticals(VRTX - Cramer's Take - Stockpickr) announced Tuesday an agreement with regulators in the U.S. and Europe that greenlights the company and partner Johnson & Johnson(JNJ - Cramer's Take - Stockpickr) to begin a new phase III study of their experimental hepatitis C drug telaprevir.
The new study will test telaprevir with standard combination therapy in 650 hepatitis C patients who could not be cured with the currently approved drugs for the disease, pegylated interferon and ribavirin. These are difficult to treat patients, including patients known as "null responders," which means they did not respond at all to previous treatments.
Vertex and Johnson & Johnson, which are co-developing telaprevir, posted an outline of this new study, dubbed REALIZE, on the ClinicalTrials.gov Web site in June. Vertex shares fell at that time because the new phase III trial suggested that an early regulatory approval filing for telaprevir in 2009 for treatment-resistant patients was not in the cards.
As it stands now, Vertex and Johnson & Johnson won't likely seek approval for telaprevir until the second half of 2010, based on data from a separate phase III study of the drug in treatment-naïve hepatitis C patients.
Vertex and its partner have previously released results from a smaller study in treatment-resistant patients that showed telaprevir to be effective in attacking the virus that causes hepatitis C.
The new phase III study announced today will test two different regimens of telaprevir in patients who previously did not respond to conventional hepatitis C treatment with pegylated interferon and ribavirin.
The study differs from previous studies in that patients will be treated with two regimens of telaprevir for a full 48 weeks instead of the accelerated 24-week treatment cycle used for treatment-naïve patients.
In addition, one of the arms of the new study will see whether a "lead-in" treatment with interferon and ribavirin alone before dosing of telaprevir may improve overall cure rates.
Vertex and its partner are locked in a tight race with Schering-Plough(SGP - Cramer's Take - Stockpickr) to be the first with a new direct antiviral drug aimed at hepatitis C approved in the U.S. and Europe.
mfg ipollit
letzte Änderungen...
- VRTX und SUPG verkauft
- OSIP deutlich reduziert
- neue Positionen in Addex, NicOx und Sucampo
22,5% Genmab http://finance.yahoo.com/q?s=GEN.CO
9,9% Onyx http://finance.yahoo.com/q?s=ONXX
9,0% Regeneron http://finance.yahoo.com/q?s=REGN
6,9% Evotec http://finance.yahoo.com/q?s=EVT.DE
5,4% Cubist http://finance.yahoo.com/q?s=CBST
5,2% Intercell http://finance.yahoo.com/q?s=IJE.F
4,9% Isis http://finance.yahoo.com/q?s=ISIS
4,7% Exelixis http://finance.yahoo.com/q?s=EXEL
4,4% Micromet http://finance.yahoo.com/q?s=MITI
4,3% Array http://finance.yahoo.com/q?s=ARRY
3,7% Arena http://finance.yahoo.com/q?s=ARNA
3,4% Addex http://finance.yahoo.com/q?s=ADXN.SW
3,4% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,0% OSI http://finance.yahoo.com/q?s=OSIP
2,4% Incyte http://finance.yahoo.com/q?s=INCY
2,4% Rigel http://finance.yahoo.com/q?s=RIGL
2,3% Sucampo http://finance.yahoo.com/q?s=SCMP
2,2% NicOx http://finance.yahoo.com/q?s=COX.PA
Watch-Liste im Moment: Cytos, Ariad, Pozen, Trubion, Curis, Medigene, Momenta
mfg ipollit
- VRTX und SUPG verkauft
- OSIP deutlich reduziert
- neue Positionen in Addex, NicOx und Sucampo
22,5% Genmab http://finance.yahoo.com/q?s=GEN.CO
9,9% Onyx http://finance.yahoo.com/q?s=ONXX
9,0% Regeneron http://finance.yahoo.com/q?s=REGN
6,9% Evotec http://finance.yahoo.com/q?s=EVT.DE
5,4% Cubist http://finance.yahoo.com/q?s=CBST
5,2% Intercell http://finance.yahoo.com/q?s=IJE.F
4,9% Isis http://finance.yahoo.com/q?s=ISIS
4,7% Exelixis http://finance.yahoo.com/q?s=EXEL
4,4% Micromet http://finance.yahoo.com/q?s=MITI
4,3% Array http://finance.yahoo.com/q?s=ARRY
3,7% Arena http://finance.yahoo.com/q?s=ARNA
3,4% Addex http://finance.yahoo.com/q?s=ADXN.SW
3,4% ViroPharma http://finance.yahoo.com/q?s=VPHM
3,0% OSI http://finance.yahoo.com/q?s=OSIP
2,4% Incyte http://finance.yahoo.com/q?s=INCY
2,4% Rigel http://finance.yahoo.com/q?s=RIGL
2,3% Sucampo http://finance.yahoo.com/q?s=SCMP
2,2% NicOx http://finance.yahoo.com/q?s=COX.PA
Watch-Liste im Moment: Cytos, Ariad, Pozen, Trubion, Curis, Medigene, Momenta
mfg ipollit
zu Intercell...
http://www.boerse-express.com/pages/691859
18.08.2008
Intercell baut für die nächsten Monate Spannung auf
Biotech-Unternehmen sitzt nach wie vor auf einem komfortablen Cashpolster
Beim Biotech-Unternehmen Intercell erwarten Anleger für das zweite Halbjahr einen regen Newsflow. So stehen die Marktzulassungen in den USA, der EU und Australien für den Impfstoff gegen Japanische Enzephalities an. Damit verbunden könnte es auch zu Meilenstein-Zahlungen von Novartis und aus einem Vertrag mit dem US-Militär kommen. Weiters könnten Nachrichten zur JEV-Zulassung in Japan kommen, auch ist für 2008 noch der Start der klinischen Phase II/IIIStudien für den Pseudomonas-Wirkstoff und der Start der Phase I-Studie für Pneumokokkus geplant.
Mit Daten zum Verlauf der Phase II-Studie des S. aureus-Impfstoffes mit Merck wird für Anfang 2009 gerechnet. "Signifikante Verzögerungen könnten unsere Bewertung negativ beeinflussen", kommentieren die Analysten von Lehman Brothers den Zeitplan.
Während sie allerdings davon ausgehen, dass ein Teil des möglicherweise anstehenden positiven Newsflows bereits im , Montag, ihre Kaufempfehlungen für den Biotech-Titel.
Die Zahlen für das zweite Quartal brachten keine grossen Überraschungen und lagen im Rahmen der Erwartungen. Ein Periodenergebnis von minus 8,65 Mio. Euro im ersten Halbjahr (nach minus 15,57 Mio. Euro) zeigt, dass Intercell am Weg in die Gewinnzone ist. Das Unternehmen bekräftigte auch das Ziel, im Gesamtjahr profitabel sein zu wollen.
Intercell sitzt zudem nach wie vor auf einem komfortablen Cash-Polster: Zum Halbjahr waren es 258 Mio. Euro, angepasst um die Cash-Zahlung für die amerikanische Iomai und weiteren 40 Mio. Euro, die im zweiten Halbjahr von Novartis fliessen könnten, sind es nach wie vor mehr als 200 Mio. Euro. (bs)
Aus dem Börse Express vom 18. August 2008
**************
http://www.stock-world.de/nachrichten/aktien/2690818-Interce…
Lichtenstein (aktiencheck.de AG) - Nach Meinung der Experten von "TradeCentre.de" ist die Aktie von Intercell (ISIN AT0000612601/ WKN A0D8HW) weiterhin aussichtsreich.
Das österreichische Biotechunternehmen wolle sich für einen Kaufpreis von gut 120 Millionen Euro die amerikanische Iomai einverleiben. Die Transaktion werde mit 1,7 Millionen Intercell-Aktien und einer Barzahlung von circa 77 Millionen Euro gestemmt. Alexander von Gabain, CSO und Mitgründer der Intercell AG, freue sich über den Deal. "Iomai passt hervorragend zu uns. Wir haben durch die Übernahme unsere Pipeline erheblich verstärkt", sage das Vorstandsmitglied im Gespräch mit "TradeCentre.de".
In der Pipeline der amerikanischen Gesellschaft werde ein Impfpflaster gegen Reisedurchfallkrankheit entwickelt. "Wir planen für diesen Impfstoff eine zulassungsrelevante klinische Phase III bereits im ersten Halbjahr des kommenden Jahres", sage der CSO. 2011 könnte das Produkt reif für die Marktzulassung sein. "Das Impfpflaster von Iomai passt perfekt zu unserem Impfstoff gegen Japanische Enzephalitis (JE)", ergänze Gabain. Zudem würden die Österreicher durch den Kauf von Iomai zwei weitere bedeutende Produkte in der klinischen Entwicklung erhalten. Beispielsweise ein Pflaster zur Vorbeugung von Influenzapandemie, welches sich in der Phase II befinde.
Das größte Highlight in diesem Jahr bei Intercell sei die Marktzulassung bei der FDA in den USA des Impfstoff gegen JE. "Wir rechnen in Kürze mit der Zulassung in den USA und im weiteren Verlauf des Jahres mit der Zulassung in Europa und Australien. Die Entwicklung liegt voll im Plan." Das Marktvolumen der beiden Reiseimpfstoffe schätze das Unternehmen auf mehr als eine Milliarde Dollar pro Jahr ein. Vertriebspartner bei JE sei übrigens die Schweizer Novartis. Bekomme Intercell grünes Licht aus den USA, würden rund 20 Millionen Euro an Meilensteinzahlungen in die Kasse der Wiener fließen. Anschließend erhalte das Unternehmen prozentuale Erlöse aus den Produktverkäufen.
Für das Geschäftsjahr würden die Experten erneut mit einem Anstieg des Umsatzes auf circa 60 Millionen Euro rechnen. Einen starken Anstieg des Profits sollten Anteilseigner des Unternehmens in diesem Jahr nicht erwarten. "Unsere Pipeline ist durch Iomai breiter und wir haben dadurch auch höhere Aufwendungen für Forschung und Entwicklung", sage Gabain. Dennoch betone der CSO, das Jahr 2008 mit einem Gewinn abzuschließen. Ziel von Intercell sei es, die Pipeline zu stärken und Innovationen weiter voranzutreiben; allerdings noch nicht den Profit zu maximieren. Cash sei nach der Übernahme mit circa 200 Millionen Euro immer noch reichlich vorhanden.
Immer wieder geistere das Gerücht über die Börse, dass Anteilseigner Novartis das Unternehmen komplett übernehmen wolle. "Wir sind sehr glücklich mit Novartis. Das beruht auf Gegenseitigkeit." Ob die Schweizer irgendwann einmal ein Übernahmeangebot machen würden, habe Gabain nicht ausschließen wollen. Derzeit würden diesbezüglich aber keine Gespräche stattfinden, so der Vorstand.
"Ich glaube, dass Novartis erst einmal abwartet, ob wir unsere Ziele mit unserer Pipeline umsetzen können. Wenn wir dies gezeigt haben, könnte Novartis durchaus Überlegungen anstrengen, uns zu übernehmen. Wir arbeiten darauf aber nicht hin. Nach Möglichkeit wollen wir eigenständig bleiben und die Genentech der Vakzine-Szene werden", fasse Gabain die Spekulationen auf eine Übernahme zusammen. In sechs bis sieben Jahren könne sich der Mitgründer von Intercell einen Umsatz von einer Milliarde Euro vorstellen. Spätestens dann sollte die Gesellschaft auch mit branchenüblichen EBIT-Margen von 30 Prozent wirtschaften.
Nach Ansicht der Experten von "TradeCentre.de" ist die Aktie des über 1,3 Milliarden Euro schweren Intercell-Konzerns unverändert aussichtsreich. (Analyse vom 21.07.2008)
mfg ipollit
http://www.boerse-express.com/pages/691859
18.08.2008
Intercell baut für die nächsten Monate Spannung auf
Biotech-Unternehmen sitzt nach wie vor auf einem komfortablen Cashpolster
Beim Biotech-Unternehmen Intercell erwarten Anleger für das zweite Halbjahr einen regen Newsflow. So stehen die Marktzulassungen in den USA, der EU und Australien für den Impfstoff gegen Japanische Enzephalities an. Damit verbunden könnte es auch zu Meilenstein-Zahlungen von Novartis und aus einem Vertrag mit dem US-Militär kommen. Weiters könnten Nachrichten zur JEV-Zulassung in Japan kommen, auch ist für 2008 noch der Start der klinischen Phase II/IIIStudien für den Pseudomonas-Wirkstoff und der Start der Phase I-Studie für Pneumokokkus geplant.
Mit Daten zum Verlauf der Phase II-Studie des S. aureus-Impfstoffes mit Merck wird für Anfang 2009 gerechnet. "Signifikante Verzögerungen könnten unsere Bewertung negativ beeinflussen", kommentieren die Analysten von Lehman Brothers den Zeitplan.
Während sie allerdings davon ausgehen, dass ein Teil des möglicherweise anstehenden positiven Newsflows bereits im , Montag, ihre Kaufempfehlungen für den Biotech-Titel.
Die Zahlen für das zweite Quartal brachten keine grossen Überraschungen und lagen im Rahmen der Erwartungen. Ein Periodenergebnis von minus 8,65 Mio. Euro im ersten Halbjahr (nach minus 15,57 Mio. Euro) zeigt, dass Intercell am Weg in die Gewinnzone ist. Das Unternehmen bekräftigte auch das Ziel, im Gesamtjahr profitabel sein zu wollen.
Intercell sitzt zudem nach wie vor auf einem komfortablen Cash-Polster: Zum Halbjahr waren es 258 Mio. Euro, angepasst um die Cash-Zahlung für die amerikanische Iomai und weiteren 40 Mio. Euro, die im zweiten Halbjahr von Novartis fliessen könnten, sind es nach wie vor mehr als 200 Mio. Euro. (bs)
Aus dem Börse Express vom 18. August 2008
**************
http://www.stock-world.de/nachrichten/aktien/2690818-Interce…
Lichtenstein (aktiencheck.de AG) - Nach Meinung der Experten von "TradeCentre.de" ist die Aktie von Intercell (ISIN AT0000612601/ WKN A0D8HW) weiterhin aussichtsreich.
Das österreichische Biotechunternehmen wolle sich für einen Kaufpreis von gut 120 Millionen Euro die amerikanische Iomai einverleiben. Die Transaktion werde mit 1,7 Millionen Intercell-Aktien und einer Barzahlung von circa 77 Millionen Euro gestemmt. Alexander von Gabain, CSO und Mitgründer der Intercell AG, freue sich über den Deal. "Iomai passt hervorragend zu uns. Wir haben durch die Übernahme unsere Pipeline erheblich verstärkt", sage das Vorstandsmitglied im Gespräch mit "TradeCentre.de".
In der Pipeline der amerikanischen Gesellschaft werde ein Impfpflaster gegen Reisedurchfallkrankheit entwickelt. "Wir planen für diesen Impfstoff eine zulassungsrelevante klinische Phase III bereits im ersten Halbjahr des kommenden Jahres", sage der CSO. 2011 könnte das Produkt reif für die Marktzulassung sein. "Das Impfpflaster von Iomai passt perfekt zu unserem Impfstoff gegen Japanische Enzephalitis (JE)", ergänze Gabain. Zudem würden die Österreicher durch den Kauf von Iomai zwei weitere bedeutende Produkte in der klinischen Entwicklung erhalten. Beispielsweise ein Pflaster zur Vorbeugung von Influenzapandemie, welches sich in der Phase II befinde.
Das größte Highlight in diesem Jahr bei Intercell sei die Marktzulassung bei der FDA in den USA des Impfstoff gegen JE. "Wir rechnen in Kürze mit der Zulassung in den USA und im weiteren Verlauf des Jahres mit der Zulassung in Europa und Australien. Die Entwicklung liegt voll im Plan." Das Marktvolumen der beiden Reiseimpfstoffe schätze das Unternehmen auf mehr als eine Milliarde Dollar pro Jahr ein. Vertriebspartner bei JE sei übrigens die Schweizer Novartis. Bekomme Intercell grünes Licht aus den USA, würden rund 20 Millionen Euro an Meilensteinzahlungen in die Kasse der Wiener fließen. Anschließend erhalte das Unternehmen prozentuale Erlöse aus den Produktverkäufen.
Für das Geschäftsjahr würden die Experten erneut mit einem Anstieg des Umsatzes auf circa 60 Millionen Euro rechnen. Einen starken Anstieg des Profits sollten Anteilseigner des Unternehmens in diesem Jahr nicht erwarten. "Unsere Pipeline ist durch Iomai breiter und wir haben dadurch auch höhere Aufwendungen für Forschung und Entwicklung", sage Gabain. Dennoch betone der CSO, das Jahr 2008 mit einem Gewinn abzuschließen. Ziel von Intercell sei es, die Pipeline zu stärken und Innovationen weiter voranzutreiben; allerdings noch nicht den Profit zu maximieren. Cash sei nach der Übernahme mit circa 200 Millionen Euro immer noch reichlich vorhanden.
Immer wieder geistere das Gerücht über die Börse, dass Anteilseigner Novartis das Unternehmen komplett übernehmen wolle. "Wir sind sehr glücklich mit Novartis. Das beruht auf Gegenseitigkeit." Ob die Schweizer irgendwann einmal ein Übernahmeangebot machen würden, habe Gabain nicht ausschließen wollen. Derzeit würden diesbezüglich aber keine Gespräche stattfinden, so der Vorstand.
"Ich glaube, dass Novartis erst einmal abwartet, ob wir unsere Ziele mit unserer Pipeline umsetzen können. Wenn wir dies gezeigt haben, könnte Novartis durchaus Überlegungen anstrengen, uns zu übernehmen. Wir arbeiten darauf aber nicht hin. Nach Möglichkeit wollen wir eigenständig bleiben und die Genentech der Vakzine-Szene werden", fasse Gabain die Spekulationen auf eine Übernahme zusammen. In sechs bis sieben Jahren könne sich der Mitgründer von Intercell einen Umsatz von einer Milliarde Euro vorstellen. Spätestens dann sollte die Gesellschaft auch mit branchenüblichen EBIT-Margen von 30 Prozent wirtschaften.
Nach Ansicht der Experten von "TradeCentre.de" ist die Aktie des über 1,3 Milliarden Euro schweren Intercell-Konzerns unverändert aussichtsreich. (Analyse vom 21.07.2008)
mfg ipollit
Antwort auf Beitrag Nr.: 34.797.510 von ipollit am 21.08.08 16:39:52ICLL...
US-Armee plant Ankauf von Impfstoff gegen Japanische Enzephalitis
21.08.2008
Wien (Österreich), 21. August 2008 - Die Intercell AG (VSE: ICLL) gab heute bekannt, dass die "Defense Logistics Agency" (DLA) des amerikanischen Verteidigungsministeriums kurzfristig eine offizielle Anfrage zur Angebotslegung (RFP, Request for Proposal) über den Ankauf des Impfstoffs gegen Japanische Enzephalitis ausgeschrieben hat. Da die Japanische Enzephalitis (JE) eine ernste und stetig wachsende Gesundheitsbedrohung für die in Asien lebenden Menschen darstellt, plant die DLA den Kauf des JE-Impfstoffs, um US-amerikanische Soldaten und Militär-Einsatzkräfte in den betroffenen Gebieten effektiv vor der Infektionskrankheit schützen zu können.
Die Kernpunkte der DLA-Ausschreibung lauten:
» Exklusiv-Vertrag zwischen DLA und dem Anbieter zur Lieferung der erforderlichen Mengen an JE-Impfstoff
» Mehrjähriger Vertrag mit einer jährlich möglichen Preis- und Mengenanpassung
» Mindestvertragsdauer von 5 Jahren
Die Intercell AG steht schon längere Zeit wegen des geplanten Auftrags in engem Kontakt mit der DLA. Intercell sieht in der Entscheidung über das Zulassungsverfahren der BLA (Biological License Application) durch die amerikanische FDA (Food and Drug Administration), die in den nächsten Monaten bevorsteht, einen wesentlichen Grund dafür, dass die offizielle Anfrage zur Angebotslegung gerade jetzt erfolgt. Intercell hat den Antrag auf Marktzulassung für den JE-Impfstoff in den USA bereits im Dezember 2007 eingereicht und erwartet innerhalb der nächsten Monate die Genehmigung durch die FDA - und damit auch die ersten möglichen Verkäufe für die US-Streitkräfte.
"Wir freuen uns, dass die DLA jetzt die konkrete Ausschreibung zur Vertragserstellung mit Intercell über die Lieferung des Impfstoffs gegen Japanische Enzephalitis bekannt gegeben hat", sagt Gerd Zettlmeissl, CEO der Intercell AG. "Der JE-Impfstoff von Intercell wurde gemeinsam mit dem amerikanischen Walter Reed Army Institute of Research entwickelt. Wir freuen uns daher, diese wertvolle Zusammenarbeit mithilfe eines langfristigen Exklusiv-Vertrags über die Verwendung des Impfstoffs im Rahmen des militärischen JE-Immunisierungsprogramms auszubauen. Intercell wird umgehend ein entsprechendes Angebot an die DLA legen und ist zuversichtlich, den Vertrag zu gegebener Zeit erfolgreich abschließen zu können."
Japanische Enzephalitis, eine von Stechmücken übertragene flavivirale und sehr ansteckende Infektion, tritt vor allem in Asien auf, breitet sich inzwischen aber auch auf Gebiete aus, die bisher nicht betroffen waren. Daher stellt der Virus eine stete gesundheitliche Gefahr für Millionen Reisende und Militärbedienstete dar, die sich in den betroffenen Gebieten, darunter auch China und Indien, aufhalten. Da es keine Behandlung für Japanische Enzephalitis gibt, ist eine Impfung die einzig mögliche Maßnahme gegen die Krankheit.
mfg ipollit
US-Armee plant Ankauf von Impfstoff gegen Japanische Enzephalitis
21.08.2008
Wien (Österreich), 21. August 2008 - Die Intercell AG (VSE: ICLL) gab heute bekannt, dass die "Defense Logistics Agency" (DLA) des amerikanischen Verteidigungsministeriums kurzfristig eine offizielle Anfrage zur Angebotslegung (RFP, Request for Proposal) über den Ankauf des Impfstoffs gegen Japanische Enzephalitis ausgeschrieben hat. Da die Japanische Enzephalitis (JE) eine ernste und stetig wachsende Gesundheitsbedrohung für die in Asien lebenden Menschen darstellt, plant die DLA den Kauf des JE-Impfstoffs, um US-amerikanische Soldaten und Militär-Einsatzkräfte in den betroffenen Gebieten effektiv vor der Infektionskrankheit schützen zu können.
Die Kernpunkte der DLA-Ausschreibung lauten:
» Exklusiv-Vertrag zwischen DLA und dem Anbieter zur Lieferung der erforderlichen Mengen an JE-Impfstoff
» Mehrjähriger Vertrag mit einer jährlich möglichen Preis- und Mengenanpassung
» Mindestvertragsdauer von 5 Jahren
Die Intercell AG steht schon längere Zeit wegen des geplanten Auftrags in engem Kontakt mit der DLA. Intercell sieht in der Entscheidung über das Zulassungsverfahren der BLA (Biological License Application) durch die amerikanische FDA (Food and Drug Administration), die in den nächsten Monaten bevorsteht, einen wesentlichen Grund dafür, dass die offizielle Anfrage zur Angebotslegung gerade jetzt erfolgt. Intercell hat den Antrag auf Marktzulassung für den JE-Impfstoff in den USA bereits im Dezember 2007 eingereicht und erwartet innerhalb der nächsten Monate die Genehmigung durch die FDA - und damit auch die ersten möglichen Verkäufe für die US-Streitkräfte.
"Wir freuen uns, dass die DLA jetzt die konkrete Ausschreibung zur Vertragserstellung mit Intercell über die Lieferung des Impfstoffs gegen Japanische Enzephalitis bekannt gegeben hat", sagt Gerd Zettlmeissl, CEO der Intercell AG. "Der JE-Impfstoff von Intercell wurde gemeinsam mit dem amerikanischen Walter Reed Army Institute of Research entwickelt. Wir freuen uns daher, diese wertvolle Zusammenarbeit mithilfe eines langfristigen Exklusiv-Vertrags über die Verwendung des Impfstoffs im Rahmen des militärischen JE-Immunisierungsprogramms auszubauen. Intercell wird umgehend ein entsprechendes Angebot an die DLA legen und ist zuversichtlich, den Vertrag zu gegebener Zeit erfolgreich abschließen zu können."
Japanische Enzephalitis, eine von Stechmücken übertragene flavivirale und sehr ansteckende Infektion, tritt vor allem in Asien auf, breitet sich inzwischen aber auch auf Gebiete aus, die bisher nicht betroffen waren. Daher stellt der Virus eine stete gesundheitliche Gefahr für Millionen Reisende und Militärbedienstete dar, die sich in den betroffenen Gebieten, darunter auch China und Indien, aufhalten. Da es keine Behandlung für Japanische Enzephalitis gibt, ist eine Impfung die einzig mögliche Maßnahme gegen die Krankheit.
mfg ipollit
Genmab... die positiven CLL-PIII Daten haben jetzt wie erwartet einen Meilenstein von 48,5 Mio USD an Genmab ausgelöst.
21.08.2008 16:27
Genmab Reaches Fifth Milestone in Ofatumumab Collaboration
COPENHAGEN, August 21 /PRNewswire-FirstCall/ -- Genmab (News) A/S (OMX: GEN) announced today it has reached the fifth milestone for ofatumumab (HuMax-CD20(R)) under the terms of its collaboration with GlaxoSmithKline (GSK). A milestone payment of approximately DKK 232.7 million (approximately USD 48.5 million) was triggered by the achievement of positive results in the Phase III study of ofatumumab in refractory chronic lymphocytic leukemia (CLL). Genmab has received an approximate total of DKK 552 million (approximately USD 110 million) in milestone payments under the collaboration so far.
"While achievement of this milestone represents an important potential turning point for Genmab," said Lisa N. Drakeman, Ph.D., Chief Executive Officer of Genmab, "it is even more important for CLL patients who are waiting for new treatments to combat their difficult to treat disease."
Ofatumumab is an investigational new generation fully human monoclonal antibody that targets a distinct, membrane proximal, small loop epitope (specific antibody binding site) of the CD20 molecule on B cells. Ofatumumab is being developed to treat CLL, follicular non-Hodgkin's lymphoma, diffuse large B-cell lymphoma, rheumatoid arthritis and relapsing remitting Multiple Sclerosis under a co-development and commercialization agreement between Genmab and GlaxoSmithKline. It is not yet approved in any country.
mfg ipollit
21.08.2008 16:27
Genmab Reaches Fifth Milestone in Ofatumumab Collaboration
COPENHAGEN, August 21 /PRNewswire-FirstCall/ -- Genmab (News) A/S (OMX: GEN) announced today it has reached the fifth milestone for ofatumumab (HuMax-CD20(R)) under the terms of its collaboration with GlaxoSmithKline (GSK). A milestone payment of approximately DKK 232.7 million (approximately USD 48.5 million) was triggered by the achievement of positive results in the Phase III study of ofatumumab in refractory chronic lymphocytic leukemia (CLL). Genmab has received an approximate total of DKK 552 million (approximately USD 110 million) in milestone payments under the collaboration so far.
"While achievement of this milestone represents an important potential turning point for Genmab," said Lisa N. Drakeman, Ph.D., Chief Executive Officer of Genmab, "it is even more important for CLL patients who are waiting for new treatments to combat their difficult to treat disease."
Ofatumumab is an investigational new generation fully human monoclonal antibody that targets a distinct, membrane proximal, small loop epitope (specific antibody binding site) of the CD20 molecule on B cells. Ofatumumab is being developed to treat CLL, follicular non-Hodgkin's lymphoma, diffuse large B-cell lymphoma, rheumatoid arthritis and relapsing remitting Multiple Sclerosis under a co-development and commercialization agreement between Genmab and GlaxoSmithKline. It is not yet approved in any country.
mfg ipollit
Micromet...
nach wie vor außer Rand und Band!
sehr schön wäre es, wenn MITI diese Kurse zu einer ausführlichen KE nutzen würde!
Micromet: Another Seal of Approval
by: Ohad Hammer posted on: August 21, 2008 | about stocks: MITI
There is always a debate regarding market efficiency and to what extent stock prices represent the available information about a company. Micromet’s (MITI) surge last week shows that in some cases, the market is far from being efficient. The spike of more than 35% in the last two trading sessions is attributed to the publication of a short article in Science Magazine, one of the world’s most prestigious scientific journals. The article contained clinical data from an ongoing phase I trial of Micromet’s lead candidate, MT103 (partnered with Medimmune). The data was spectacular, showing a strong, dose dependent response in concert with a good safety profile, exactly the kind of data that can put a small biotech in the spotlight. Ironically, the article contained data which has already been presented more than two months ago at the ICML in Switzerland.
Regardless of whether market reaction was justified, publishing clinical data at such an early stage in Science should be viewed as an indication for the scientific community’s embrace of Micromet and its BiTE platform. The BiTE platform relies on monoclonal antibodies for stimulating the patient’s immune system to attack cancer cells that have managed to evade or suppress the body’s immune response. Although most attention is given to the first product from the platform, MT103, it can generate an unlimited number of agents against a variety of cancers, making it a potential revolution in the way cancer is treated.
It still remains to be seen whether additional BiTE agents will be as promising as MT103, but examination of the clinical data leads to the conclusion that Micromet now has one of the most exciting technologies in the biotech industry. The BiTE platform represents a truly novel class of anti-cancer agents and is not like anything else out there. It facilitates the construction of bi-specific antibodies that simultaneously bind cancer cells and the body’s most potent immune cells (T cells), leading to a strong, long lasting and escalating immune response against tumors. In the past decades, there have been numerous attempts to create effective bi-specific antibodies, all of which failed. One exception may be a Biotest’s HRS3/A9, which showed promising efficacy but could not be produced in sufficient quantities for further clinical evaluations. The area of bi-specific antibodies has been long abandoned by the antibody industry and MT103 can be seen as the long anticipated breakthrough that overcame most of the hurdles. It seems that Micromet found the ideal formula for generating a potent yet safe anti-tumor response with bi-specific antibodies.
BiTE antibodies are small enough to get T- cells within sufficient proximity to cancer cells and unleash their lethal mechanisms in a targeted and specific manner. Their small size may also help them penetrate areas inaccessible for other agents. In addition, because the actual attack is done by the patient’s most potent immune cells, which can proliferate and multiply upon activation, it takes a tiny amount of BiTE antibodies for generating a systemic response. This may explain the incredibly low doses of MT103 that led to clinical responses in the phase I clinical trial. When compared to other antibodies and chemotherapeutic agents on a normalized basis, MT103 is certainly one of the most potent compounds to have ever been tested in humans, if not the most potent one.
Promising Data
The Science article reported data for thirty eight NHL (non-Hodgkin lymphoma) patients, who received various doses of MT103. The trial focused on two subtypes of NHL, follicular lymphoma [FL] and Mantle cell lymphoma [MCL], which together represent around 35% of the NHL market. There were 11 cases of objective response (four complete and seven partial), which appeared primarily in the cohorts who received the higher doses. Strikingly, all seven patients who received the highest dose responded to the treatment, two of whom had a complete response. As of the latest follow up (June 1st) all these responses were ongoing for a period of 1-8.5 months, on top of one response in a patient who had received a lower dose that was ongoing for more than a year. The issue of response duration is cardinal because many treatments, especially Rituxan containing regimens manage to achieve long lasting responses of more than a year in similar patient populations, so in order to be regarded as a viable alternative, MT103 must keep patients in remission for substantial periods of time.
It is important to understand that the significance of the data is not necessarily in the commercial opportunity of MT103 for these patient populations, which fortunately have a slower disease onset and enjoy an abundance of treatment options. The challenging competitive landscape leads to high entry barriers for new therapies for FL and MCL patients. Furthermore, there are additional antibodies in development that bind the same target MT103 binds, CD19. The most advanced of these agents is Sanofi-Aventis’ (SNY) SAR3419, based on Immunogen’s (IMGN) technology, which is currently in two phase I trials. Additional CD19 targeting agents are being developed preclinically in the hands of Genentech (DNA), Seattle Genetics (SGEN), Medarex (MEDX) and Xencor. MT103’s results are actually the first validation of CD19 as a target and are therefore likely to motivate these companies to advance their candidates into the clinic as soon as possible.
Hopefully, MT103 will find its way to the NHL market as the next line of defense for certain subsets of patients, but the real potential lies somewhere else. Because the BiTE platform is a universal and modular platform, it could be utilized to generate a plethora of candidates for indications with limited treatment options, primarily solid tumors. Solid tumors, such as lung and breast cancers are less sensitive to available treatments due to poor accessibility and are consequently responsible for over 90% of cancer related deaths worldwide. BiTE antibodies may have a competitive edge over other therapies due to their small size and certain properties of the immune cells they activate. They may also be advantageous for targets that cannot be hit by other antibody-based platforms, because they do not require internalization in order to be effective. For more on the mechanism by which BiTE antibodies operate and why they are considered so promising, click here. The first BiTE antibody for solid tumors, MT110, recently entered the clinic, and is expected to generate initial data next year.
Substantial Risks
Investors should be aware of the long list of risks associated with MT103 in particular and the BiTE platform in general. With respect to MT103, the data is preliminary and based on a limited number of patients, so it may turn out to be less effective in larger trials. In addition, the NHL market is full of promising compounds, some of which demonstrated comparable or even better efficacy in similar patient populations. A closer look at patient demographics reveals that patients in the highest cohort were younger and less heavily pretreated compared to patients in lower doses, which might explain the 100% response rate.
Despite the explosive potential of the BiTE platform, there is the obvious unpredictability associated with early stage drug development. There is no guarantee that additional BiTE antibodies will replicate MT103’s success, and even if some do, it might be after multiple failures, like is often the case with novel disrupting technologies.
Safety is also a concern, especially in therapies that manipulate the immune system, and until a compound is evaluated in large enough populations the fear of unexpected adverse events is always there. This explains Micromet’s decision not to escalate the dose in the MT103’s trial further, despite the fact that the maximum tolerated dose [MTD] was not reached. Because MT103 is the first product of the BiTE platform, safety issues can lead to delays or even jeopardize the entire platform. Therefore, when a dose escalation study reaches a sufficiently active dose, there is much more to lose from increased toxicity than to gain from better efficacy. In the case of solid tumors, however, it will probably take dose levels near or at the MTD in order to achieve the needed efficacy, so the risk there is obviously higher.
Three Main Events
For the remainder of the year, there will be three important events for Micromet.
Most importantly, the company will publish additional data in December at the Annual Society of Hematology [ASH] annual meeting. The data set will include an update from the ongoing trial in NHL patients as well as preliminary results from a phase II study that evaluates MT103 in Acute lymphoblastic leukemia [ALL]. Choosing ALL as a second indication makes sense because it is estimated that over 90% of ALL cases express CD19.
The data from the NHL trial is very important not only because it will include additional patients and an update on the durability of the responses, but also because data from Sanofi-Aventis’ SAR3419 will be presented. This will give investors the opportunity to assess the BiTE platform relatively to competing platforms.
The ALL study has important implications as well, because it represents an attractive route for commercialization for MT103. ALL is an aggressive blood cancer with a dismal prognosis and very few treatment options, in contrast to the NHL subtypes in the phase I trial. If MT103 demonstrates sufficient activity against ALL, this indication represents a faster route to market, with very little competition upon approval. In addition, the ALL study is very significant for the future development of MT103, because it represents a different treatment modality. The study does not evaluate MT103 as a primary treatment, but as consolidation in patients who receive chemotherapy but still have leukemia remnants in their bone marrow. The company views this study as a probe for the utility of MT103 as a secondary treatment as well as for the potential of the compound for other aggressive blood cancers.
The second event, which is supposed to take place before ASH, is rumored to be a partnership deal for one of Micromet’s pre-clinical programs or a technology licensing deal with a major pharmaceutical company. Such a development will serve as another validation of Micromet’s potential, provide highly needed cash infusion and decrease the overall risk.
The third event will be some sort of a capital raising deal, which will probably happen sooner rather than later given the recent stock movement. This may put some pressure on the stock in the coming months.
I have been getting a lot of questions from investors with respect to Micromet’s target price. My answer is that the huge potential of the company warrants holding it as long as the BiTE platform continues to produce impressive clinical results. Nevertheless, with a market cap of $220M and a year to date return of over 170%, Micromet is not a cheap stock. The recent jump following the Science article, coupled with the anticipated round of financing might imply that now is a good time to take some gains off the table with the purpose of waiting for a lower entry point down the road, prior to the ASH annual meeting.
Disclosure: Author is Long MITI, IMGN, SGEN
mfg ipollit
nach wie vor außer Rand und Band!
sehr schön wäre es, wenn MITI diese Kurse zu einer ausführlichen KE nutzen würde!
Micromet: Another Seal of Approval
by: Ohad Hammer posted on: August 21, 2008 | about stocks: MITI
There is always a debate regarding market efficiency and to what extent stock prices represent the available information about a company. Micromet’s (MITI) surge last week shows that in some cases, the market is far from being efficient. The spike of more than 35% in the last two trading sessions is attributed to the publication of a short article in Science Magazine, one of the world’s most prestigious scientific journals. The article contained clinical data from an ongoing phase I trial of Micromet’s lead candidate, MT103 (partnered with Medimmune). The data was spectacular, showing a strong, dose dependent response in concert with a good safety profile, exactly the kind of data that can put a small biotech in the spotlight. Ironically, the article contained data which has already been presented more than two months ago at the ICML in Switzerland.
Regardless of whether market reaction was justified, publishing clinical data at such an early stage in Science should be viewed as an indication for the scientific community’s embrace of Micromet and its BiTE platform. The BiTE platform relies on monoclonal antibodies for stimulating the patient’s immune system to attack cancer cells that have managed to evade or suppress the body’s immune response. Although most attention is given to the first product from the platform, MT103, it can generate an unlimited number of agents against a variety of cancers, making it a potential revolution in the way cancer is treated.
It still remains to be seen whether additional BiTE agents will be as promising as MT103, but examination of the clinical data leads to the conclusion that Micromet now has one of the most exciting technologies in the biotech industry. The BiTE platform represents a truly novel class of anti-cancer agents and is not like anything else out there. It facilitates the construction of bi-specific antibodies that simultaneously bind cancer cells and the body’s most potent immune cells (T cells), leading to a strong, long lasting and escalating immune response against tumors. In the past decades, there have been numerous attempts to create effective bi-specific antibodies, all of which failed. One exception may be a Biotest’s HRS3/A9, which showed promising efficacy but could not be produced in sufficient quantities for further clinical evaluations. The area of bi-specific antibodies has been long abandoned by the antibody industry and MT103 can be seen as the long anticipated breakthrough that overcame most of the hurdles. It seems that Micromet found the ideal formula for generating a potent yet safe anti-tumor response with bi-specific antibodies.
BiTE antibodies are small enough to get T- cells within sufficient proximity to cancer cells and unleash their lethal mechanisms in a targeted and specific manner. Their small size may also help them penetrate areas inaccessible for other agents. In addition, because the actual attack is done by the patient’s most potent immune cells, which can proliferate and multiply upon activation, it takes a tiny amount of BiTE antibodies for generating a systemic response. This may explain the incredibly low doses of MT103 that led to clinical responses in the phase I clinical trial. When compared to other antibodies and chemotherapeutic agents on a normalized basis, MT103 is certainly one of the most potent compounds to have ever been tested in humans, if not the most potent one.
Promising Data
The Science article reported data for thirty eight NHL (non-Hodgkin lymphoma) patients, who received various doses of MT103. The trial focused on two subtypes of NHL, follicular lymphoma [FL] and Mantle cell lymphoma [MCL], which together represent around 35% of the NHL market. There were 11 cases of objective response (four complete and seven partial), which appeared primarily in the cohorts who received the higher doses. Strikingly, all seven patients who received the highest dose responded to the treatment, two of whom had a complete response. As of the latest follow up (June 1st) all these responses were ongoing for a period of 1-8.5 months, on top of one response in a patient who had received a lower dose that was ongoing for more than a year. The issue of response duration is cardinal because many treatments, especially Rituxan containing regimens manage to achieve long lasting responses of more than a year in similar patient populations, so in order to be regarded as a viable alternative, MT103 must keep patients in remission for substantial periods of time.
It is important to understand that the significance of the data is not necessarily in the commercial opportunity of MT103 for these patient populations, which fortunately have a slower disease onset and enjoy an abundance of treatment options. The challenging competitive landscape leads to high entry barriers for new therapies for FL and MCL patients. Furthermore, there are additional antibodies in development that bind the same target MT103 binds, CD19. The most advanced of these agents is Sanofi-Aventis’ (SNY) SAR3419, based on Immunogen’s (IMGN) technology, which is currently in two phase I trials. Additional CD19 targeting agents are being developed preclinically in the hands of Genentech (DNA), Seattle Genetics (SGEN), Medarex (MEDX) and Xencor. MT103’s results are actually the first validation of CD19 as a target and are therefore likely to motivate these companies to advance their candidates into the clinic as soon as possible.
Hopefully, MT103 will find its way to the NHL market as the next line of defense for certain subsets of patients, but the real potential lies somewhere else. Because the BiTE platform is a universal and modular platform, it could be utilized to generate a plethora of candidates for indications with limited treatment options, primarily solid tumors. Solid tumors, such as lung and breast cancers are less sensitive to available treatments due to poor accessibility and are consequently responsible for over 90% of cancer related deaths worldwide. BiTE antibodies may have a competitive edge over other therapies due to their small size and certain properties of the immune cells they activate. They may also be advantageous for targets that cannot be hit by other antibody-based platforms, because they do not require internalization in order to be effective. For more on the mechanism by which BiTE antibodies operate and why they are considered so promising, click here. The first BiTE antibody for solid tumors, MT110, recently entered the clinic, and is expected to generate initial data next year.
Substantial Risks
Investors should be aware of the long list of risks associated with MT103 in particular and the BiTE platform in general. With respect to MT103, the data is preliminary and based on a limited number of patients, so it may turn out to be less effective in larger trials. In addition, the NHL market is full of promising compounds, some of which demonstrated comparable or even better efficacy in similar patient populations. A closer look at patient demographics reveals that patients in the highest cohort were younger and less heavily pretreated compared to patients in lower doses, which might explain the 100% response rate.
Despite the explosive potential of the BiTE platform, there is the obvious unpredictability associated with early stage drug development. There is no guarantee that additional BiTE antibodies will replicate MT103’s success, and even if some do, it might be after multiple failures, like is often the case with novel disrupting technologies.
Safety is also a concern, especially in therapies that manipulate the immune system, and until a compound is evaluated in large enough populations the fear of unexpected adverse events is always there. This explains Micromet’s decision not to escalate the dose in the MT103’s trial further, despite the fact that the maximum tolerated dose [MTD] was not reached. Because MT103 is the first product of the BiTE platform, safety issues can lead to delays or even jeopardize the entire platform. Therefore, when a dose escalation study reaches a sufficiently active dose, there is much more to lose from increased toxicity than to gain from better efficacy. In the case of solid tumors, however, it will probably take dose levels near or at the MTD in order to achieve the needed efficacy, so the risk there is obviously higher.
Three Main Events
For the remainder of the year, there will be three important events for Micromet.
Most importantly, the company will publish additional data in December at the Annual Society of Hematology [ASH] annual meeting. The data set will include an update from the ongoing trial in NHL patients as well as preliminary results from a phase II study that evaluates MT103 in Acute lymphoblastic leukemia [ALL]. Choosing ALL as a second indication makes sense because it is estimated that over 90% of ALL cases express CD19.
The data from the NHL trial is very important not only because it will include additional patients and an update on the durability of the responses, but also because data from Sanofi-Aventis’ SAR3419 will be presented. This will give investors the opportunity to assess the BiTE platform relatively to competing platforms.
The ALL study has important implications as well, because it represents an attractive route for commercialization for MT103. ALL is an aggressive blood cancer with a dismal prognosis and very few treatment options, in contrast to the NHL subtypes in the phase I trial. If MT103 demonstrates sufficient activity against ALL, this indication represents a faster route to market, with very little competition upon approval. In addition, the ALL study is very significant for the future development of MT103, because it represents a different treatment modality. The study does not evaluate MT103 as a primary treatment, but as consolidation in patients who receive chemotherapy but still have leukemia remnants in their bone marrow. The company views this study as a probe for the utility of MT103 as a secondary treatment as well as for the potential of the compound for other aggressive blood cancers.
The second event, which is supposed to take place before ASH, is rumored to be a partnership deal for one of Micromet’s pre-clinical programs or a technology licensing deal with a major pharmaceutical company. Such a development will serve as another validation of Micromet’s potential, provide highly needed cash infusion and decrease the overall risk.
The third event will be some sort of a capital raising deal, which will probably happen sooner rather than later given the recent stock movement. This may put some pressure on the stock in the coming months.
I have been getting a lot of questions from investors with respect to Micromet’s target price. My answer is that the huge potential of the company warrants holding it as long as the BiTE platform continues to produce impressive clinical results. Nevertheless, with a market cap of $220M and a year to date return of over 170%, Micromet is not a cheap stock. The recent jump following the Science article, coupled with the anticipated round of financing might imply that now is a good time to take some gains off the table with the purpose of waiting for a lower entry point down the road, prior to the ASH annual meeting.
Disclosure: Author is Long MITI, IMGN, SGEN
mfg ipollit
Antwort auf Beitrag Nr.: 34.797.943 von ipollit am 21.08.08 17:08:03hier noch der zugehörige Link... http://seekingalpha.com/article/91907-micromet-another-seal-…
Antwort auf Beitrag Nr.: 34.744.942 von ipollit am 17.08.08 19:30:51hier auch noch mal der Barron's Artikel...
http://www.smartmoney.com/barrons/index.cfm?story=20080820-b…
Today From Barron's
Browsing Among Biotechs
By Lawrence C. Strauss |Published: August 20, 2008
THE OUTCOME OF Bristol-Myers Squibb's (BMY: 21.94, +0.42, +1.95%) $4.5 billion bid for the portion of ImClone Systems that it doesn't already own remains uncertain. So is the conclusion of Roche Holding's $44 billion offer for the shares of Genentech that it doesn't possess. Both targets want the bids raised.
But one thing is certain: Mergers, buyouts and takeovers involving biotech outfits will become more common as Big Pharma increasingly tries to fatten its product pipeline by acquiring proven, as well as promising, bioengineered drugs.
For Bristol, the big prize is Erbitux, an ImClone (IMCL: 64.05, -0.01, -0.01%) cancer treatment. For Roche (RO.Switzerland), the lure is a raft of Genentech (DNA: 97.50, -0.49, -0.50%) products, ranging from Activase, a heart-attack treatment, to Avastin, a cancer drug, to Nutropin, a growth-hormone product.
Driving the need for new products are the coming expirations of patents on several important drugs owned by Big Pharma. The end of patent protection, of course, opens the market to cheaper generic versions of the drugs.
In the not-very-distant future, the mother of all expirations will hit Lipitor, Pfizer's (PFE: 19.39, +0.11, +0.57%) wildly successful cholesterol drug, which had more than $12 billion in annual global sales last year. Lipitor goes off-patent in 2011. Other blockbuster drugs that are losing their patent protection include Wyeth's (WYE: 42.00, -0.14, -0.33%) Effexor (depression and anxiety), 2010; Merck's (MRK: 34.81, -0.13, -0.37%) Singulair (allergies), 2012; and Eli Lilly's (LLY: 47.15, -0.26, -0.54%) Zyprexa (schizophrenia), 2011.
Many of the large drug manufacturers "have three or four products accounting for the bulk of earnings," notes Christopher Schott, a pharmaceutical analyst at JPMorgan.
At the same time, these companies' costly research-and-development efforts have had mixed results. Despite Big Pharma's spending billions and billions, says Jay Markowitz, a T. Rowe Price health-care analyst, "a number of companies are facing a significant patent cliff, where billions in revenues are going to disappear."
Many large pharmaceutical outfits, however, have a lot of cash on their balance sheets to make acquisitions. Factor in the dollar's weakness, which makes U.S. companies look particularly enticing to foreigners, and the biotechnology/pharmaceuticals market looks particularly ripe for merger-and-acquisition deals.
"The natural synergy is for a company to acquire a company they already know well and have a partnership with on a key drug," says Steven Silver, an analyst at Standard & Poor's.
For example, Genentech, which last week rejected Roche's $89-a-share proposal as too low, has a long history of dealings with the Swiss pharmaceutical giant, which has about a 56% stake in the San Francisco biotech firm and markets some of its products.
"The larger pharma companies want to have total control over the drugs in the pipelines," says Frank Sustersic, a portfolio manager at Turner Investment Partners. In 2007, for example, Eli Lilly acquired Icos, with which it had a joint venture on Cialis, an erectile-dysfunction drug. It paid $2.3 billion for the acquisition.
"It looks as if pharma is going after the companies that have existing products," which by definition have obtained regulatory approval, says Vinay Thapar, a senior investment analyst at American Century Investments. (Larger companies will continue to do licensing agreements in which they pay a smaller company to help develop a drug, in exchange for a cut of future profits, says Thapar. In such cases, the larger entity doesn't typically acquire the smaller one.)
Thapar and other analysts point to Onyx Pharmaceuticals (ONXX: 41.74, -0.35, -0.83%) as a potential target of Bayer (BAY. Germany). Onyx has a joint venture with the German pharmaceutical giant for Nexavar, which is used to treat kidney and liver cancers.
"At some point, you could see Bayer wanting to control 100% of the assets," says Sustersic, who thinks that Bayer might pay as much as $65 per share. Nexavar is believed to have more upside, especially as it secures approval in other markets (it was recently given the green light by China as a liver-cancer treatment), and even more if it can be used for other cancers, including those of the breast, lungs and skin.
One argument for buying existing drugs — rather than developing new ones — is the grueling U.S. Food and Drug Administration testing that new products face.
"The FDA is perceived to have swung more to the safety side in the balance between safety and efficacy," says Markowitz. That means that getting drugs through the regulatory process is taking longer, although it's probably good for consumers. Adds Thapar: "The FDA is becoming increasingly difficult to handicap."
Not all of the focus is on products already in the market, however. Vertex Pharmaceuticals (VRTX: 26.75, +0.43, +1.63%), another company that some investors view as an eventual takeover candidate, is developing Telaprevir, a substance that promises to reduce the time needed to treat hepatitis-C, a potentially fatal liver ailment. Telaprevir is currently in stage III testing, the last phase of the clinical- trial process. Vertex has worked with Johnson & Johnson (JNJ: 71.31, +0.23, +0.32%) in developing that drug.
Vertex's shares sold off recently after Schering-Plough (SGP: 19.86, -0.60, -2.93%) announced that it had good results with a possible competitor to Telaprevir. Still, some analysts think that the Vertex drug shouldn't be underestimated. "In my opinion, it will be the first direct antiviral drug for hepatitis-C to hit the market and meaningfully improve patient outcomes," says T. Rowe Price's Markowitz.
Another company that could spark an acquirer's interest is Amylin Pharmaceuticals (AMLN: 26.69, -0.22, -0.81%). It's working on a Type 2 diabetes drug that could be injected once a week instead of daily. Amylin has several development partners, including Eli Lilly, on the drug. It already markets two diabetes drugs, Symlin and Byetta. But some analysts say the product under development could boast major advantages. "A once-weekly drug that lowers glucose substantially, induces weight loss, isn't associated with hypoglycemia, and lacks a cardiovascular safety signal has multibillion-dollar potential," says Markowitz.
Anyone scanning the biotech ranks for potential acquisition targets shouldn't overlook United Therapeutics (UTHR: 104.36, -0.32, -0.30%). It has one characteristic that lots of other small biotechs would envy: It's in the black. One of its most promising products is Remodulin, which is used to treat hypertension in the blood vessels of the lungs. Although there are other PAH drugs, Turner Investment Partners' Sustersic says that United Therapeutics is "one of the clear leaders, and they potentially could have a big blockbuster."
American Century's Thapar is similarly impressed by Alexion Pharmaceuticals (ALXN: 88.30, -1.44, -1.60%), which earned six cents a share in its most recent quarter, versus a loss of 75 cents a share a year earlier. Its drugs include Soliris, which treats a rare disorder called paroxysmal nocturnal hemoglobinuria, which destroys red blood cells.
At $389,000 a year per patient, Soliris is hugely expensive — but it targets a clearly defined patient base.
The drug had net sales of nearly $60 million in the second quarter, up from $45.5 million in the first quarter and $9.8 million a year earlier. Regulators have approved its use in the U.S. and Europe, and it's expected to be introduced in Japan toward the end of next year. Alexion retains distribution rights in the U.S. and overseas, says Sustersic, who maintains that Soliris sales could reach at least $500 million a year.
Sounds like a decent prescription for a bigger company seeking a revenue boost.
mfg ipollit
http://www.smartmoney.com/barrons/index.cfm?story=20080820-b…
Today From Barron's
Browsing Among Biotechs
By Lawrence C. Strauss |Published: August 20, 2008
THE OUTCOME OF Bristol-Myers Squibb's (BMY: 21.94, +0.42, +1.95%) $4.5 billion bid for the portion of ImClone Systems that it doesn't already own remains uncertain. So is the conclusion of Roche Holding's $44 billion offer for the shares of Genentech that it doesn't possess. Both targets want the bids raised.
But one thing is certain: Mergers, buyouts and takeovers involving biotech outfits will become more common as Big Pharma increasingly tries to fatten its product pipeline by acquiring proven, as well as promising, bioengineered drugs.
For Bristol, the big prize is Erbitux, an ImClone (IMCL: 64.05, -0.01, -0.01%) cancer treatment. For Roche (RO.Switzerland), the lure is a raft of Genentech (DNA: 97.50, -0.49, -0.50%) products, ranging from Activase, a heart-attack treatment, to Avastin, a cancer drug, to Nutropin, a growth-hormone product.
Driving the need for new products are the coming expirations of patents on several important drugs owned by Big Pharma. The end of patent protection, of course, opens the market to cheaper generic versions of the drugs.
In the not-very-distant future, the mother of all expirations will hit Lipitor, Pfizer's (PFE: 19.39, +0.11, +0.57%) wildly successful cholesterol drug, which had more than $12 billion in annual global sales last year. Lipitor goes off-patent in 2011. Other blockbuster drugs that are losing their patent protection include Wyeth's (WYE: 42.00, -0.14, -0.33%) Effexor (depression and anxiety), 2010; Merck's (MRK: 34.81, -0.13, -0.37%) Singulair (allergies), 2012; and Eli Lilly's (LLY: 47.15, -0.26, -0.54%) Zyprexa (schizophrenia), 2011.
Many of the large drug manufacturers "have three or four products accounting for the bulk of earnings," notes Christopher Schott, a pharmaceutical analyst at JPMorgan.
At the same time, these companies' costly research-and-development efforts have had mixed results. Despite Big Pharma's spending billions and billions, says Jay Markowitz, a T. Rowe Price health-care analyst, "a number of companies are facing a significant patent cliff, where billions in revenues are going to disappear."
Many large pharmaceutical outfits, however, have a lot of cash on their balance sheets to make acquisitions. Factor in the dollar's weakness, which makes U.S. companies look particularly enticing to foreigners, and the biotechnology/pharmaceuticals market looks particularly ripe for merger-and-acquisition deals.
"The natural synergy is for a company to acquire a company they already know well and have a partnership with on a key drug," says Steven Silver, an analyst at Standard & Poor's.
For example, Genentech, which last week rejected Roche's $89-a-share proposal as too low, has a long history of dealings with the Swiss pharmaceutical giant, which has about a 56% stake in the San Francisco biotech firm and markets some of its products.
"The larger pharma companies want to have total control over the drugs in the pipelines," says Frank Sustersic, a portfolio manager at Turner Investment Partners. In 2007, for example, Eli Lilly acquired Icos, with which it had a joint venture on Cialis, an erectile-dysfunction drug. It paid $2.3 billion for the acquisition.
"It looks as if pharma is going after the companies that have existing products," which by definition have obtained regulatory approval, says Vinay Thapar, a senior investment analyst at American Century Investments. (Larger companies will continue to do licensing agreements in which they pay a smaller company to help develop a drug, in exchange for a cut of future profits, says Thapar. In such cases, the larger entity doesn't typically acquire the smaller one.)
Thapar and other analysts point to Onyx Pharmaceuticals (ONXX: 41.74, -0.35, -0.83%) as a potential target of Bayer (BAY. Germany). Onyx has a joint venture with the German pharmaceutical giant for Nexavar, which is used to treat kidney and liver cancers.
"At some point, you could see Bayer wanting to control 100% of the assets," says Sustersic, who thinks that Bayer might pay as much as $65 per share. Nexavar is believed to have more upside, especially as it secures approval in other markets (it was recently given the green light by China as a liver-cancer treatment), and even more if it can be used for other cancers, including those of the breast, lungs and skin.
One argument for buying existing drugs — rather than developing new ones — is the grueling U.S. Food and Drug Administration testing that new products face.
"The FDA is perceived to have swung more to the safety side in the balance between safety and efficacy," says Markowitz. That means that getting drugs through the regulatory process is taking longer, although it's probably good for consumers. Adds Thapar: "The FDA is becoming increasingly difficult to handicap."
Not all of the focus is on products already in the market, however. Vertex Pharmaceuticals (VRTX: 26.75, +0.43, +1.63%), another company that some investors view as an eventual takeover candidate, is developing Telaprevir, a substance that promises to reduce the time needed to treat hepatitis-C, a potentially fatal liver ailment. Telaprevir is currently in stage III testing, the last phase of the clinical- trial process. Vertex has worked with Johnson & Johnson (JNJ: 71.31, +0.23, +0.32%) in developing that drug.
Vertex's shares sold off recently after Schering-Plough (SGP: 19.86, -0.60, -2.93%) announced that it had good results with a possible competitor to Telaprevir. Still, some analysts think that the Vertex drug shouldn't be underestimated. "In my opinion, it will be the first direct antiviral drug for hepatitis-C to hit the market and meaningfully improve patient outcomes," says T. Rowe Price's Markowitz.
Another company that could spark an acquirer's interest is Amylin Pharmaceuticals (AMLN: 26.69, -0.22, -0.81%). It's working on a Type 2 diabetes drug that could be injected once a week instead of daily. Amylin has several development partners, including Eli Lilly, on the drug. It already markets two diabetes drugs, Symlin and Byetta. But some analysts say the product under development could boast major advantages. "A once-weekly drug that lowers glucose substantially, induces weight loss, isn't associated with hypoglycemia, and lacks a cardiovascular safety signal has multibillion-dollar potential," says Markowitz.
Anyone scanning the biotech ranks for potential acquisition targets shouldn't overlook United Therapeutics (UTHR: 104.36, -0.32, -0.30%). It has one characteristic that lots of other small biotechs would envy: It's in the black. One of its most promising products is Remodulin, which is used to treat hypertension in the blood vessels of the lungs. Although there are other PAH drugs, Turner Investment Partners' Sustersic says that United Therapeutics is "one of the clear leaders, and they potentially could have a big blockbuster."
American Century's Thapar is similarly impressed by Alexion Pharmaceuticals (ALXN: 88.30, -1.44, -1.60%), which earned six cents a share in its most recent quarter, versus a loss of 75 cents a share a year earlier. Its drugs include Soliris, which treats a rare disorder called paroxysmal nocturnal hemoglobinuria, which destroys red blood cells.
At $389,000 a year per patient, Soliris is hugely expensive — but it targets a clearly defined patient base.
The drug had net sales of nearly $60 million in the second quarter, up from $45.5 million in the first quarter and $9.8 million a year earlier. Regulators have approved its use in the U.S. and Europe, and it's expected to be introduced in Japan toward the end of next year. Alexion retains distribution rights in the U.S. and overseas, says Sustersic, who maintains that Soliris sales could reach at least $500 million a year.
Sounds like a decent prescription for a bigger company seeking a revenue boost.
mfg ipollit
Antwort auf Beitrag Nr.: 34.801.283 von ipollit am 21.08.08 22:06:45hier ein älterer Artikel, der zeigt, wie dringend die Pharmas neue Medikamente brauchen, weil ihre Blockbuster in den nächsten Jahren ihren Patent-Schutz verlieren.
Unten jeweils der Umsatzanteil des Unternehmens der alleine zwischen 2010-2012 wegbrechen dürfte.
http://www.forbes.com/home/business/2007/01/19/pfizer-pharma…
Big Pharma's Black Hole
Matthew Herper, 01.22.07, 8:30 AM ET
Big Pharma is heading "off a cliff" and into "a black hole," according to Wall Street.
Analysts are already using such big scary metaphors to describe the challenges facing the drug industry in five years, when drug makers will face the worst series of patent expirations ever.
Between 2010 and 2011, Big Pharma will lose 28% of their current sales, according to pharmaceutical analyst James Kelly of Goldman Sachs--who is calling this period "the patent black hole." Starting in 2008 and going through 2011, Kelly predicts annualized sales growth of only 2% for big drug makers.
Timothy Anderson of Prudential Equity Group says that this generic "cliff" is the largest in the history of pharma. Most of the companies he covers, including Pfizer (nyse: PFE - news - people ), Merck (nyse: MRK - news - people ), GlaxoSmithKline (nyse: GSK - news - people ), and Eli Lilly (nyse: LLY - news - people ), will lose top-sellers between 2010 and 2013. Already, he says, the loss of blockbuster products like Pfizer\'s Lipitor for high cholesterol, the world\'s best-selling drug, and Eli Lilly\'s Zyprexa for schizophrenia, is shaping decision-making at big drug makers.
A case in point: Pfizer, which is expected to announce deep job cuts and cost-cutting measures on Monday when it holds an analyst meeting in New York. The world\'s largest drug firm\'s new chief executive, Jeffrey Kindler, has said he wants to "transform" the company and took the unprecedented step of cutting some 2,000 jobs from the drug giant\'s 10,000-strong U.S. sales force. But that was before torcetrapib, a cholesterol drug that Pfizer had bet on as a Lipitor replacement, failed catastrophically in a big clinical trial.
Monday morning, Pfizer announced that its sales increased 2% in 2006. Earnings increased dramatically partly because of the sale of Pfizer\'s consumer business to Johnson & Johnson (nyse: JNJ - news - people ). But so-called operating earnings--those tracked by analysts--dropped in the fourth quarter by 12%, to 43 cents per share. That figure still tops the average forecast of Wall Street analysts polled by Thomson Financial. Further announcements on job cuts are expected later in the day.
In a note to investors last Monday, Barbara Ryan at Deutsche Bank predicted that Kindler will cut 8,000 more jobs from sales, manufacturing, and research and save $2 billion on top of the $4 billion worth of cost cuts he has already announced. That money, she says, could help Pfizer make a big acquisition to make up for the Lipitor loss. According to Anderson\'s estimates, Pfizer will lose 41% of its sales between 2010 and 2012, more than any drug maker he covers except for much smaller Forest Laboratories (nyse: FRX - news - people ).
Both Ryan and Kelly say it is worth betting on Kindler\'s planned transformation, especially since Pfizer pays a healthy dividend that was hiked by 21% this year. Anderson prefers Schering-Plough (nyse: SGP - news - people ), which has less generic exposure, and Novartis (nyse: NVS - news - people ), which has actually made a big investment in generics.
The problem for Pfizer and its peers is not just that older drugs are going off patent, but that new ones are not making up the difference. Last year, only 26 medicines or vaccines were approved by the Food and Drug Administration, half as many as in 1996, the year that Lipitor was approved. And although Pfizer launched an innovative stop-smoking pill, Chantix, and a cancer-fighter, Sutent, those medicines are not expected to make up for the losses of big sellers like antidepressant Zoloft and hypertension pill Norvasc, not to mention the $12-billion-a-year Lipitor.
Drug Patent Decimation 2010
According to Prudential Equity Group's Timothy Anderson, these companies face big losses to generics between 2010 and 2012.
Drug Patents Expiring
Total Company Sales Expiring
Forest Laboratories
Namenda, Lexapro
86%
Pfizer
Aricept, Lipitor, Viagra, Detrol, Geodon
41%
AstraZeneca
Arimidex, Seroquel, Symbicort
38%
Bristol-Myers Squibb
Plavix, Avapro, Abilify
30%
GlaxoSmithKline
Advair, Avandia
23%
Eli Lilly
Zyprexa
22%
Merck
Cozaar/Hyzaar, Singulair
22%
Wyeth
Effexor, Protonix
22%
Novartis
Femara, Diovan
14%
Roche
None
None
Schering-Plough
None
None
Source: Prudential Equity Group
"On one side I feel very sorry for the industry, on the other side it is a great opportunity for our generics business," says Daniel Vasella, chief executive of Novartis. Overall, he predicts that the pharma business "will remain a growth industry, the demand remains and demand will accelerate" due to the aging of the population. However, as more people take pills, governments will also do more to ratchet down drug prices. Right now, Congress is debating measures that might reduce the price Medicare pays for drugs, for instance.
On Monday, then, the entire drug industry will be watching to see what Jeff Kindler, the Pfizer chief executive, does. In losing torcetrapib, which he himself had called one of the biggest drug opportunities in years, he is coming off a dramatic failure. It seems certain that now Pfizer has to make plans to defend its turf and increase its earnings power. A few good new drugs wouldn\'t hurt either. The question at hand: Can drug companies, which have thrived based on the research successes of the mid-1990s, bring forth a new series of blockbuster medicines?
Goldman Sachs analyst Kelly, who coined the "black hole" term, sees a good chance for some drug makers. Aside from Pfizer, he likes Eli Lilly, which is developing a new blood thinner to compete with Plavix, the second-best-selling pill in the world made by Sanofi-Aventis (nyse: SNY - news - people ) and Bristol-Myers Squibb (nyse: BMY - news - people ). But he says that data emerging on new medicines over the next two years will be crucial to determining what kind of damage drug makers sustain five years from now--and how many more jobs these embattled giants will have to cut.[/i]
mfg ipollit
Unten jeweils der Umsatzanteil des Unternehmens der alleine zwischen 2010-2012 wegbrechen dürfte.
http://www.forbes.com/home/business/2007/01/19/pfizer-pharma…
Big Pharma's Black Hole
Matthew Herper, 01.22.07, 8:30 AM ET
Big Pharma is heading "off a cliff" and into "a black hole," according to Wall Street.
Analysts are already using such big scary metaphors to describe the challenges facing the drug industry in five years, when drug makers will face the worst series of patent expirations ever.
Between 2010 and 2011, Big Pharma will lose 28% of their current sales, according to pharmaceutical analyst James Kelly of Goldman Sachs--who is calling this period "the patent black hole." Starting in 2008 and going through 2011, Kelly predicts annualized sales growth of only 2% for big drug makers.
Timothy Anderson of Prudential Equity Group says that this generic "cliff" is the largest in the history of pharma. Most of the companies he covers, including Pfizer (nyse: PFE - news - people ), Merck (nyse: MRK - news - people ), GlaxoSmithKline (nyse: GSK - news - people ), and Eli Lilly (nyse: LLY - news - people ), will lose top-sellers between 2010 and 2013. Already, he says, the loss of blockbuster products like Pfizer\'s Lipitor for high cholesterol, the world\'s best-selling drug, and Eli Lilly\'s Zyprexa for schizophrenia, is shaping decision-making at big drug makers.
A case in point: Pfizer, which is expected to announce deep job cuts and cost-cutting measures on Monday when it holds an analyst meeting in New York. The world\'s largest drug firm\'s new chief executive, Jeffrey Kindler, has said he wants to "transform" the company and took the unprecedented step of cutting some 2,000 jobs from the drug giant\'s 10,000-strong U.S. sales force. But that was before torcetrapib, a cholesterol drug that Pfizer had bet on as a Lipitor replacement, failed catastrophically in a big clinical trial.
Monday morning, Pfizer announced that its sales increased 2% in 2006. Earnings increased dramatically partly because of the sale of Pfizer\'s consumer business to Johnson & Johnson (nyse: JNJ - news - people ). But so-called operating earnings--those tracked by analysts--dropped in the fourth quarter by 12%, to 43 cents per share. That figure still tops the average forecast of Wall Street analysts polled by Thomson Financial. Further announcements on job cuts are expected later in the day.
In a note to investors last Monday, Barbara Ryan at Deutsche Bank predicted that Kindler will cut 8,000 more jobs from sales, manufacturing, and research and save $2 billion on top of the $4 billion worth of cost cuts he has already announced. That money, she says, could help Pfizer make a big acquisition to make up for the Lipitor loss. According to Anderson\'s estimates, Pfizer will lose 41% of its sales between 2010 and 2012, more than any drug maker he covers except for much smaller Forest Laboratories (nyse: FRX - news - people ).
Both Ryan and Kelly say it is worth betting on Kindler\'s planned transformation, especially since Pfizer pays a healthy dividend that was hiked by 21% this year. Anderson prefers Schering-Plough (nyse: SGP - news - people ), which has less generic exposure, and Novartis (nyse: NVS - news - people ), which has actually made a big investment in generics.
The problem for Pfizer and its peers is not just that older drugs are going off patent, but that new ones are not making up the difference. Last year, only 26 medicines or vaccines were approved by the Food and Drug Administration, half as many as in 1996, the year that Lipitor was approved. And although Pfizer launched an innovative stop-smoking pill, Chantix, and a cancer-fighter, Sutent, those medicines are not expected to make up for the losses of big sellers like antidepressant Zoloft and hypertension pill Norvasc, not to mention the $12-billion-a-year Lipitor.
Drug Patent Decimation 2010
According to Prudential Equity Group's Timothy Anderson, these companies face big losses to generics between 2010 and 2012.
Drug Patents Expiring
Total Company Sales Expiring
Forest Laboratories
Namenda, Lexapro
86%
Pfizer
Aricept, Lipitor, Viagra, Detrol, Geodon
41%
AstraZeneca
Arimidex, Seroquel, Symbicort
38%
Bristol-Myers Squibb
Plavix, Avapro, Abilify
30%
GlaxoSmithKline
Advair, Avandia
23%
Eli Lilly
Zyprexa
22%
Merck
Cozaar/Hyzaar, Singulair
22%
Wyeth
Effexor, Protonix
22%
Novartis
Femara, Diovan
14%
Roche
None
None
Schering-Plough
None
None
Source: Prudential Equity Group
"On one side I feel very sorry for the industry, on the other side it is a great opportunity for our generics business," says Daniel Vasella, chief executive of Novartis. Overall, he predicts that the pharma business "will remain a growth industry, the demand remains and demand will accelerate" due to the aging of the population. However, as more people take pills, governments will also do more to ratchet down drug prices. Right now, Congress is debating measures that might reduce the price Medicare pays for drugs, for instance.
On Monday, then, the entire drug industry will be watching to see what Jeff Kindler, the Pfizer chief executive, does. In losing torcetrapib, which he himself had called one of the biggest drug opportunities in years, he is coming off a dramatic failure. It seems certain that now Pfizer has to make plans to defend its turf and increase its earnings power. A few good new drugs wouldn\'t hurt either. The question at hand: Can drug companies, which have thrived based on the research successes of the mid-1990s, bring forth a new series of blockbuster medicines?
Goldman Sachs analyst Kelly, who coined the "black hole" term, sees a good chance for some drug makers. Aside from Pfizer, he likes Eli Lilly, which is developing a new blood thinner to compete with Plavix, the second-best-selling pill in the world made by Sanofi-Aventis (nyse: SNY - news - people ) and Bristol-Myers Squibb (nyse: BMY - news - people ). But he says that data emerging on new medicines over the next two years will be crucial to determining what kind of damage drug makers sustain five years from now--and how many more jobs these embattled giants will have to cut.[/i]
mfg ipollit
Antwort auf Beitrag Nr.: 34.797.510 von ipollit am 21.08.08 16:39:52Intercell kaufen
22.08.2008
Erste Bank
Wien (aktiencheck.de AG) - Für die Analystin der Erste Bank, Vladimira Urbankova, ist die Aktie von Intercell (ISIN AT0000612601 / WKN A0D8HW) unverändert eine Kaufempfehlung.
Das führende österreichische Biotech-Unternehmen habe diese Woche die HJ-Zahlen 2008 bekannt gegeben. Der Umsatz sei - wie von den Analysten erwartet - deutlich auf EUR 17,6 Mio. gesteigert worden, wobei hier auch Teile der Zahlungen aus der Novartis-Kooperation enthalten seien. Der Nettoverlust habe deutlich verringert werden können und im 1. Halbjahr EUR 8,6 Mio. betragen. Die Cash-Position der Gesellschaft habe per 30.06. EUR 258,3 Mio. betragen und sei damit sehr solide.
Weiters sehr wichtig sei die Bestätigung gewesen, dass für das Gesamtjahr 2008 - trotz der kürzlichen Übernahme der US-Firma Iomai - ein positives Ergebnis erwartet werde. Dies stütze sich auf weitere erwartete Meilenstein-Zahlungen bzw. auch auf erste Umsätze aus dem Japan Enzephalitis Impfstoff mit dem US-Militär.
Für die Analysten habe sich durch die HJ-Ergebnisse nicht allzu viel Neues ergeben, da ja auch die Zahlen großteils im Rahmen ihrer Erwartungen gelegen hätten. Sie würden somit nun auf die endgültigen Marktzulassungen des JE-Impfstoffs bzw. auf weitere neue Testergebnisse bzw. Kooperationen warten. Das US-Verteidigungsministerium habe diese Woche eine offizielle Anfrage zur Angebotslegung (RFP) über den geplanten Ankauf des Intercell-Impfstoffs gegen Japanische Enzephalitis ausgeschrieben. Es handle sich um einen Exklusiv-Vertrag mit einer Mindestvertragsdauer von fünf Jahren.
Die Analysten der Erste Bank bestätigen jedenfalls ihre Kaufempfehlung für die Intercell-Aktie. Intercell sei weder von der Finanzkrise betroffen (wenn überhaupt, dann eher positiv durch höhere Zinsen auf den Geldmittelbestand), noch in irgendeiner Weise von der Konjunktur abhängig. Das aktuelle Kursziel der Analysten auf Sicht von zwölf Monaten betrage EUR 40,5. (Analyse vom 22.08.2008)
mfg ipollit
22.08.2008
Erste Bank
Wien (aktiencheck.de AG) - Für die Analystin der Erste Bank, Vladimira Urbankova, ist die Aktie von Intercell (ISIN AT0000612601 / WKN A0D8HW) unverändert eine Kaufempfehlung.
Das führende österreichische Biotech-Unternehmen habe diese Woche die HJ-Zahlen 2008 bekannt gegeben. Der Umsatz sei - wie von den Analysten erwartet - deutlich auf EUR 17,6 Mio. gesteigert worden, wobei hier auch Teile der Zahlungen aus der Novartis-Kooperation enthalten seien. Der Nettoverlust habe deutlich verringert werden können und im 1. Halbjahr EUR 8,6 Mio. betragen. Die Cash-Position der Gesellschaft habe per 30.06. EUR 258,3 Mio. betragen und sei damit sehr solide.
Weiters sehr wichtig sei die Bestätigung gewesen, dass für das Gesamtjahr 2008 - trotz der kürzlichen Übernahme der US-Firma Iomai - ein positives Ergebnis erwartet werde. Dies stütze sich auf weitere erwartete Meilenstein-Zahlungen bzw. auch auf erste Umsätze aus dem Japan Enzephalitis Impfstoff mit dem US-Militär.
Für die Analysten habe sich durch die HJ-Ergebnisse nicht allzu viel Neues ergeben, da ja auch die Zahlen großteils im Rahmen ihrer Erwartungen gelegen hätten. Sie würden somit nun auf die endgültigen Marktzulassungen des JE-Impfstoffs bzw. auf weitere neue Testergebnisse bzw. Kooperationen warten. Das US-Verteidigungsministerium habe diese Woche eine offizielle Anfrage zur Angebotslegung (RFP) über den geplanten Ankauf des Intercell-Impfstoffs gegen Japanische Enzephalitis ausgeschrieben. Es handle sich um einen Exklusiv-Vertrag mit einer Mindestvertragsdauer von fünf Jahren.
Die Analysten der Erste Bank bestätigen jedenfalls ihre Kaufempfehlung für die Intercell-Aktie. Intercell sei weder von der Finanzkrise betroffen (wenn überhaupt, dann eher positiv durch höhere Zinsen auf den Geldmittelbestand), noch in irgendeiner Weise von der Konjunktur abhängig. Das aktuelle Kursziel der Analysten auf Sicht von zwölf Monaten betrage EUR 40,5. (Analyse vom 22.08.2008)
mfg ipollit
Antwort auf Beitrag Nr.: 34.801.283 von ipollit am 21.08.08 22:06:45nochmal zu Micromet und den BI-AKs, die das Immunsystem gegen Krebs aktivieren sollen...
http://www.biotechnologie.de/bio/generator/Navigation/Deutsc…
Neuartige Immuntherapie gegen Krebs
22.08.2008
Im Kampf gegen Krebs gibt es unterschiedliche Ansätze. Einer besteht darin, das körpereigene Immunsystehm so aufzurüsten, dass es sich gegen die Tumorzellen selbst zur Wehr setzen kann. Deutsche Forscher der aus der Ludwig-Maximilians-Universität ausgegründeten Biotech-Firma Micromet haben sich diesem Ziel verschrieben und können jetzt einen ersten Erfolg verbuchen. Wie sie gemeinsam mit Kollegen aus den Universitätskliniken in Würzburg, Essen, Ulm und Mainz im Fachmagazin Science (2008, Vol. 321, S. 974 – 977) berichten, hat ein neuartiger, vom Unternehmen entwickelter Antikörper in einer ersten Studie an Patienten mit Lymphdrüsenkrebs hoffnungsvolle Ergebnisse gezeigt. Auf alle bislang verfügbaren Medikamente hatten die Betroffenen nicht mehr angesprochen, die neue Therapie hingegen wirkte.

Die Immuntherapie basiert auf Antikörpern mit zwei Greifarmen, die sowohl an die T-Zelle als auch an die Krebszelle binden können. Quelle: Micromet
Es ist eine alte Idee der Medizin, das Immunsystem im Kampf gegen Krebs aufmarschieren zu lassen und sich auf die körpereigenen Heilungskräfte zu stützen. Eigentlich kann der menschliche Organismus nämlich Krebszellen attackieren, insbesondere im Anfangsstadium der Erkrankung. Doch die Attacke ist nicht effektiv genug, irgendwann übernehmen die Krebszellen die Kontrolle. „Meiner Ansicht nach muss sich die Menschheit nur deshalb mit Krebserkankungen herumschlagen, weil die Killerzellen des Immunsystems irgendwann die Kontrolle über die Tumore verlieren“, so Patrick Bäuerle, Forschungsvorstand von Micromet, in der Süddeutschen Zeitung (2008, 16. August, Ressort Wissen).
Im Jahr 1993 wurde die Firma aus dem Institut für Immunologie der Ludwig-Maximilians-Universität München ausgegründet – angestoßen vom Institutsleiter Gerd Riethmüller, der an Immuntherapien gegen Krebs auf der Basis seiner Kenntnisse zu Mikrometastasen gearbeitet hat. Diese schon in einem sehr frühen Stadium der Krankheit ausgesäten Zellen haben der Firma auch ihren Namen gegeben. Die Geschichte des Unternehmens hat bereits einige Höhen und Tiefen erlebt, zuletzt machte Micromet 2006 mit einem Reverse Merger Schlagzeilen. Anstatt in Europa an die Börse zu gehen, haben die Münchner die bereits an der Nasdaq notierte US-amerikanische CancerVax Corp. als Hülle übernommen. Damit wurde die Firmenzentrale der neuen Micromet Inc. in die USA verlegt, Forschung und Entwicklung findet aber nach wie vor in München statt.
Wirkungsvolle Antikörper mit zwei Greifarmen
Die Entwicklung von sogenannten bispezifischen Antikörpern hat Micromet seit Ende der 90er Jahre verfolgt. Diese ganz neue Medikamentenklasse verfügt über zwei Molekülarme und ist ein kompliziertes gentechnisches Konstrukt auf der Basis von Einzelketten-Antikörpern. Normalerweise können Antikörper nur an eine Art von Molekülen binden, die Münchner Forscher haben ihm jedoch mithilfe ihrer hauseigenen BiTe-Technologie zwei Greifarme verpasst, die jeweils unterschiedliche Moleküle binden können. Der eine dockt an Rezeptoren an, die vermehrt auf der Oberfläche von bestimmten Krebszellen vorkommen (CD19). Der andere Arm bindet an einen Rezeptor (CD3) auf bestimmten weißen Blutkörperchen, den T-Zellen, die ein Teil der körpereigenen Immunabwehr darstellen.
Durch die Verbindung zwischen Krebs- und T-Zelle wird letztere ohne weitere stimulierende Faktoren derart aktiviert, dass sie die Tumorzellen auflösen kann – die Immunabwehr wird also angeregt, sich selbst gegen den Krebs zu wehren. Die Entwicklung dieses Ansatzes hat einige Jahre in Anspruch genommen. Erst 2006 konnten die Wissenschaftler klären, wie die durch die Antikörper aktivierten T-Zellen die Krebszellen effektiv zerstören können (Molecular Immunology, 2006, Vol. 43, S. 763-771). Demnach schaffen es die bispezifischen Antikörper, dass die T-Zellen den Krebszellen eine Art tödlichen Kuss geben – über den toxische Substanzen in die Tumorzellen eindringen können, was zu deren Vernichtung führt. „Wir können die T-Zellen auf den Krebs abrichten“, erläuterte Bäuerle das Potential des neuen Wirkstoffs jüngst in der Financial Times Deutschland (FTD, 2008, 18. August). „Wenn sie mit dem Antikörper verknüpft werden, macht sie das richtig wild.“ Ohne den Antikörper würden die körpereigenen Killerzellen die Krebszellen regelrecht übersehen, mithilfe des Wirkstoffs erhalten sie eine Brille und packen zu.

Die Immuntherapie ist offenbar angeschlagen, wie die Bilder aus dem Knochenmark (oben) sowie aus der Leber (unten) vor (links) und nach der Behandlung (rechts) zeigen.In der oberen Reihe sind die Krebszellen in blau, in der unteren in braun zu sehen.Quelle: Bargou et al, Science, 2008
Hoffnungsvoller erster Praxistest bei Patienten mit Lymphdrüsenkrebs
Ob der Ansatz tatsächlich auch in der Praxis funktioniert, ist nicht immer selbstverständlich, wie der Fall Tegenero gezeigt hat (mehr…). Bei den Forschern von Micromet sieht es jedoch besser aus. Wie sie gemeinsam mit Kollegen aus Universitätskliniken in Würzburg, Essen, Ulm und Mainz im Fachmagazin Science (2008, Vol. 321, S. 974 – 977) berichten, haben die bispezifischen Antikörper in einer ersten klinischen Studie hoffnungsvolle Resultate gezeigt. Insgesamt wurden 38 Patienten mit Diagnose Lymphdrüsenkrebs (Non-Hodgkin-Lymphom) mit verschiedenen Dosen des Wirkstoffs behandelt. Die Betroffenen hatten auf bislang verfügbare Medikamente (Rituxan) und Chemotherapien nicht mehr angesprochen. Die Ergebnisse sind vielversprechend. Bei allen sieben Patienten, die letztlich mit der für weitere Studien ausgewählten Dosis behandelt wurden, ist die Krankheit zurückgegangen, so die Wissenschaftler in Science. Eine solch hohe Ansprechrate ist selten – und wohl auch ein Grund dafür, dass ein so angesehenes Fachblatt wie Science frühe Studienergebnisse eines Krebswirkstoffes veröffentlicht. Nichtsdestotrotz mussten die Studienteilnehmer zum Teil hohe Nebenwirkungen über sich ergehen lassen. Eine Immuntherapie ist immer auch eine hohe Belastung. Wenn T-Zellen aktiviert werden, dann sind ähnliche Symptome wie bei einer schweren Infektion im Gange. Jahrelang wurde deshalb nach der richtigen Dosis gesucht. "Man kann innerhalb von Minuten richtig krank werden", erläutert Georg Maschmeyer, Chefarzt am Potsdamer Klinikum Ernst von Bergmann in der FTD, der vor zehn Jahren bereits erste Versuche begleitet hat.
Und so vielversprechend die Ergebnisse auch sind, noch befinden sich die Wissenschaftler von Micromet ganz am Anfang. Bis der Wirkstoff mit dem Namen Blinatumomab, für den US-Firma MedImmune (jetzt Teil des britischen Pharmakonzern AstraZeneca) als Entwicklungspartner die teuren klinischen Studien finanziert, tatsächlich den Markt erreicht, werden noch Jahre vergehen. Bis dahin muss sich der Wirkstoff auch in großen Studien mit deutlich mehr Patienten beweisen - und hier bleiben oft auch hoffnungsvolle Kandidaten auf der Strecke. Anfang 2008 hat Micromet eine Studie der Phase II mit Patienten gestartet, die an Akuter lymphatischer Leukämie (ALL) leiden. Derzeit ist die Stimmung optimistisch. „Ich bin nach diesen Daten so mutig zu sagen, dass wir eine Chance haben“, sagte Bäuerle gegenüber der Süddeutschen Zeitung.
mfg ipollit
http://www.biotechnologie.de/bio/generator/Navigation/Deutsc…
Neuartige Immuntherapie gegen Krebs
22.08.2008
Im Kampf gegen Krebs gibt es unterschiedliche Ansätze. Einer besteht darin, das körpereigene Immunsystehm so aufzurüsten, dass es sich gegen die Tumorzellen selbst zur Wehr setzen kann. Deutsche Forscher der aus der Ludwig-Maximilians-Universität ausgegründeten Biotech-Firma Micromet haben sich diesem Ziel verschrieben und können jetzt einen ersten Erfolg verbuchen. Wie sie gemeinsam mit Kollegen aus den Universitätskliniken in Würzburg, Essen, Ulm und Mainz im Fachmagazin Science (2008, Vol. 321, S. 974 – 977) berichten, hat ein neuartiger, vom Unternehmen entwickelter Antikörper in einer ersten Studie an Patienten mit Lymphdrüsenkrebs hoffnungsvolle Ergebnisse gezeigt. Auf alle bislang verfügbaren Medikamente hatten die Betroffenen nicht mehr angesprochen, die neue Therapie hingegen wirkte.
Die Immuntherapie basiert auf Antikörpern mit zwei Greifarmen, die sowohl an die T-Zelle als auch an die Krebszelle binden können. Quelle: Micromet
Es ist eine alte Idee der Medizin, das Immunsystem im Kampf gegen Krebs aufmarschieren zu lassen und sich auf die körpereigenen Heilungskräfte zu stützen. Eigentlich kann der menschliche Organismus nämlich Krebszellen attackieren, insbesondere im Anfangsstadium der Erkrankung. Doch die Attacke ist nicht effektiv genug, irgendwann übernehmen die Krebszellen die Kontrolle. „Meiner Ansicht nach muss sich die Menschheit nur deshalb mit Krebserkankungen herumschlagen, weil die Killerzellen des Immunsystems irgendwann die Kontrolle über die Tumore verlieren“, so Patrick Bäuerle, Forschungsvorstand von Micromet, in der Süddeutschen Zeitung (2008, 16. August, Ressort Wissen).
Im Jahr 1993 wurde die Firma aus dem Institut für Immunologie der Ludwig-Maximilians-Universität München ausgegründet – angestoßen vom Institutsleiter Gerd Riethmüller, der an Immuntherapien gegen Krebs auf der Basis seiner Kenntnisse zu Mikrometastasen gearbeitet hat. Diese schon in einem sehr frühen Stadium der Krankheit ausgesäten Zellen haben der Firma auch ihren Namen gegeben. Die Geschichte des Unternehmens hat bereits einige Höhen und Tiefen erlebt, zuletzt machte Micromet 2006 mit einem Reverse Merger Schlagzeilen. Anstatt in Europa an die Börse zu gehen, haben die Münchner die bereits an der Nasdaq notierte US-amerikanische CancerVax Corp. als Hülle übernommen. Damit wurde die Firmenzentrale der neuen Micromet Inc. in die USA verlegt, Forschung und Entwicklung findet aber nach wie vor in München statt.
Wirkungsvolle Antikörper mit zwei Greifarmen
Die Entwicklung von sogenannten bispezifischen Antikörpern hat Micromet seit Ende der 90er Jahre verfolgt. Diese ganz neue Medikamentenklasse verfügt über zwei Molekülarme und ist ein kompliziertes gentechnisches Konstrukt auf der Basis von Einzelketten-Antikörpern. Normalerweise können Antikörper nur an eine Art von Molekülen binden, die Münchner Forscher haben ihm jedoch mithilfe ihrer hauseigenen BiTe-Technologie zwei Greifarme verpasst, die jeweils unterschiedliche Moleküle binden können. Der eine dockt an Rezeptoren an, die vermehrt auf der Oberfläche von bestimmten Krebszellen vorkommen (CD19). Der andere Arm bindet an einen Rezeptor (CD3) auf bestimmten weißen Blutkörperchen, den T-Zellen, die ein Teil der körpereigenen Immunabwehr darstellen.
Durch die Verbindung zwischen Krebs- und T-Zelle wird letztere ohne weitere stimulierende Faktoren derart aktiviert, dass sie die Tumorzellen auflösen kann – die Immunabwehr wird also angeregt, sich selbst gegen den Krebs zu wehren. Die Entwicklung dieses Ansatzes hat einige Jahre in Anspruch genommen. Erst 2006 konnten die Wissenschaftler klären, wie die durch die Antikörper aktivierten T-Zellen die Krebszellen effektiv zerstören können (Molecular Immunology, 2006, Vol. 43, S. 763-771). Demnach schaffen es die bispezifischen Antikörper, dass die T-Zellen den Krebszellen eine Art tödlichen Kuss geben – über den toxische Substanzen in die Tumorzellen eindringen können, was zu deren Vernichtung führt. „Wir können die T-Zellen auf den Krebs abrichten“, erläuterte Bäuerle das Potential des neuen Wirkstoffs jüngst in der Financial Times Deutschland (FTD, 2008, 18. August). „Wenn sie mit dem Antikörper verknüpft werden, macht sie das richtig wild.“ Ohne den Antikörper würden die körpereigenen Killerzellen die Krebszellen regelrecht übersehen, mithilfe des Wirkstoffs erhalten sie eine Brille und packen zu.
Die Immuntherapie ist offenbar angeschlagen, wie die Bilder aus dem Knochenmark (oben) sowie aus der Leber (unten) vor (links) und nach der Behandlung (rechts) zeigen.In der oberen Reihe sind die Krebszellen in blau, in der unteren in braun zu sehen.Quelle: Bargou et al, Science, 2008
Hoffnungsvoller erster Praxistest bei Patienten mit Lymphdrüsenkrebs
Ob der Ansatz tatsächlich auch in der Praxis funktioniert, ist nicht immer selbstverständlich, wie der Fall Tegenero gezeigt hat (mehr…). Bei den Forschern von Micromet sieht es jedoch besser aus. Wie sie gemeinsam mit Kollegen aus Universitätskliniken in Würzburg, Essen, Ulm und Mainz im Fachmagazin Science (2008, Vol. 321, S. 974 – 977) berichten, haben die bispezifischen Antikörper in einer ersten klinischen Studie hoffnungsvolle Resultate gezeigt. Insgesamt wurden 38 Patienten mit Diagnose Lymphdrüsenkrebs (Non-Hodgkin-Lymphom) mit verschiedenen Dosen des Wirkstoffs behandelt. Die Betroffenen hatten auf bislang verfügbare Medikamente (Rituxan) und Chemotherapien nicht mehr angesprochen. Die Ergebnisse sind vielversprechend. Bei allen sieben Patienten, die letztlich mit der für weitere Studien ausgewählten Dosis behandelt wurden, ist die Krankheit zurückgegangen, so die Wissenschaftler in Science. Eine solch hohe Ansprechrate ist selten – und wohl auch ein Grund dafür, dass ein so angesehenes Fachblatt wie Science frühe Studienergebnisse eines Krebswirkstoffes veröffentlicht. Nichtsdestotrotz mussten die Studienteilnehmer zum Teil hohe Nebenwirkungen über sich ergehen lassen. Eine Immuntherapie ist immer auch eine hohe Belastung. Wenn T-Zellen aktiviert werden, dann sind ähnliche Symptome wie bei einer schweren Infektion im Gange. Jahrelang wurde deshalb nach der richtigen Dosis gesucht. "Man kann innerhalb von Minuten richtig krank werden", erläutert Georg Maschmeyer, Chefarzt am Potsdamer Klinikum Ernst von Bergmann in der FTD, der vor zehn Jahren bereits erste Versuche begleitet hat.
Und so vielversprechend die Ergebnisse auch sind, noch befinden sich die Wissenschaftler von Micromet ganz am Anfang. Bis der Wirkstoff mit dem Namen Blinatumomab, für den US-Firma MedImmune (jetzt Teil des britischen Pharmakonzern AstraZeneca) als Entwicklungspartner die teuren klinischen Studien finanziert, tatsächlich den Markt erreicht, werden noch Jahre vergehen. Bis dahin muss sich der Wirkstoff auch in großen Studien mit deutlich mehr Patienten beweisen - und hier bleiben oft auch hoffnungsvolle Kandidaten auf der Strecke. Anfang 2008 hat Micromet eine Studie der Phase II mit Patienten gestartet, die an Akuter lymphatischer Leukämie (ALL) leiden. Derzeit ist die Stimmung optimistisch. „Ich bin nach diesen Daten so mutig zu sagen, dass wir eine Chance haben“, sagte Bäuerle gegenüber der Süddeutschen Zeitung.
mfg ipollit
Antwort auf Beitrag Nr.: 34.825.489 von ipollit am 23.08.08 13:55:10MITI... Cash 22,4 Mio USD... Cashburn im ersten Halbjahr ca. 5 Mio USD. - eher kurzfristig als langfristig sollte Micromet die eigenen Cash-Reserven wieder auffüllen. Das aktuell erhöhte Bewertungsniveau und die positive Stimmung bieten eine gute Gelegenheit.
07.08.2008 13:04
Micromet, Inc. Reports Second Quarter 2008 Financial Results
BETHESDA, Md., Aug. 7 /PRNewswire-FirstCall/ -- Micromet, (News) Inc. , a biopharmaceutical company focusing on the development of novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced its financial results for the second quarter and six months ended June 30, 2008.
Summary of Recent Events:
In June, Micromet and MedImmune presented a clinical update for the BiTE(R) antibody blinatumomab (MT103/MEDI-538) at the International Conference on Malignant Lymphoma in Lugano, Switzerland. All seven patients with relapsed non-Hodgkin's lymphoma treated with blinatumomab at the highest dose level presented at the conference responded to the treatment with partial or complete responses. Responses also appeared to be durable. The most frequent side effects observed so far were lymphopenia, pyrexia and leukopenia. Less common adverse events included transient neutropenia and thrombocytopenia, transient increase of liver enzymes and central nervous system events, all of which were fully reversible. In addition, Micromet and MedImmune commenced treatment of patients with acute lymphoblastic leukemia in a phase 2 clinical trial of blinatumomab.
Also in June, Micromet received a milestone payment of $775,000 from Nycomed in connection with the initiation of formal preclinical safety studies for antibody MT203, which has potential applications in the treatment of inflammatory and autoimmune diseases.
In April, Micromet announced the initiation of the first phase 1 clinical trial with its BiTE antibody MT110. The study will explore the safety, pharmacokinetics, pharmacodynamics and anti-tumor activity of MT110 in patients with lung cancer and patients with gastrointestinal cancer. MT110 targets the epithelial cell adhesion molecule (EpCAM or CD326), which is highly expressed on colon, lung, breast, prostate, ovarian, gastric and pancreatic cancer cells and on cancer stem cells of colon, breast, prostate and pancreas cancers. Cancer stem cells are believed to cause metastases and recurrence of these cancers.
Also in April, Micromet presented five posters at the American Association for Cancer Research (AACR) showing recent progress on the company's proprietary BiTE antibody platform and new BiTE antibodies:
-- Anti-cancer antibodies in marketed products Herceptin(R), Erbitux(R) and Vectibix(R) and the anti-asthma antibody in Xolair(R) were successfully converted to highly potent BiTE antibodies.
-- Animal data from two studies indicated feasibility of subcutaneous administration of BiTE antibodies MT103 and MT110.
-- Animal data provided proof of concept for a BiTE antibody targeting CD33 with potential use in the treatment of acute myelogenic leukemia (AML), and a BiTE antibody targeting MCSP with potential use in the treatment of melanoma.
-- Animal data suggested a therapeutic window for a BiTE antibody targeting EpCAM in a relevant animal species.
Summarizing the events, Christian Itin, Ph.D., President and Chief Executive Officer of Micromet said: "We have made significant progress with the BiTE antibody platform introducing new BiTE antibodies that target a wide range of tumor indications and converting currently marketed therapeutic antibodies into potent BiTE antibodies with much increased activity against tumor cells. We believe that the progress on our proprietary BiTE antibody platform will allow us to expand our own product pipeline, while at the same time engaging in new collaborations on selected BiTE antibody programs."
Financial Results:
Quarter Ended June 30, 2008
For the three months ended June 30, 2008, Micromet recognized total revenues of $8.5 million, compared to $3.1 million for the same period in 2007. Total operating expenses were $14.4 million for the three months ended June 30, 2008, compared to $11.1 million for the same period in 2007. For the three months ended June 30, 2008, Micromet reported a net loss of $8.6 million, or $0.21 per basic and diluted common share, compared to a net loss of $6.5 million, or $0.20 per basic and diluted common share, for the same period in 2007.
Six Months Ended June 30, 2008
For the six months ended June 30, 2008, Micromet recognized total revenues of $14.4 million, compared to $5.8 million for the same period in 2007. Total operating expenses were $27.6 million for the six months ended June 30, 2008, compared to $21.4 million for the same period in 2007. For the six months ended June 30, 2008, Micromet reported a net loss of $14.5 million, or $0.36 per basic and diluted common share, compared to a net loss of $14.1 million, or $0.44 per basic and diluted common share, for the same period in 2007.
Micromet's cash and cash equivalents were $22.4 million as of June 30, 2008. Net cash used in operating activities was $4.7 million for the six months ended June 30, 2008 compared to $7.9 million used in operating activities for the same period in 2007.
2008 Outlook:
-- At the annual meeting of the European Society for Molecular Oncology (ESMO) in September in Stockholm, Sweden, Micromet will present data from the ongoing phase 1b clinical trial of patients with metastatic breast cancer treated with adecatumumab (MT201) in combination with docetaxel. Further, Micromet plans to initiate a phase 2 clinical trial later in 2008 to evaluate adecatumumab in an adjuvant setting.
-- At the annual meeting of the American Society for Hematology (ASH) in December, Micromet expects to provide a further update on the currently ongoing phase 1 clinical trial of blinatumomab in patients with non-Hodgkins lymphoma.
-- Also at ASH, Micromet expects to present initial results on the currently ongoing phase 2 clinical trial of blinatumomab in patients with acute lymphoblastic leukemia (ALL).
-- Finally, Micromet expects that its collaboration partner MedImmune will begin a phase 1 clinical trial in the United States evaluating blinatumomab in patients with chronic lymphocytic leukemia by the end of 2008.
mfg ipollit
07.08.2008 13:04
Micromet, Inc. Reports Second Quarter 2008 Financial Results
BETHESDA, Md., Aug. 7 /PRNewswire-FirstCall/ -- Micromet, (News) Inc. , a biopharmaceutical company focusing on the development of novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced its financial results for the second quarter and six months ended June 30, 2008.
Summary of Recent Events:
In June, Micromet and MedImmune presented a clinical update for the BiTE(R) antibody blinatumomab (MT103/MEDI-538) at the International Conference on Malignant Lymphoma in Lugano, Switzerland. All seven patients with relapsed non-Hodgkin's lymphoma treated with blinatumomab at the highest dose level presented at the conference responded to the treatment with partial or complete responses. Responses also appeared to be durable. The most frequent side effects observed so far were lymphopenia, pyrexia and leukopenia. Less common adverse events included transient neutropenia and thrombocytopenia, transient increase of liver enzymes and central nervous system events, all of which were fully reversible. In addition, Micromet and MedImmune commenced treatment of patients with acute lymphoblastic leukemia in a phase 2 clinical trial of blinatumomab.
Also in June, Micromet received a milestone payment of $775,000 from Nycomed in connection with the initiation of formal preclinical safety studies for antibody MT203, which has potential applications in the treatment of inflammatory and autoimmune diseases.
In April, Micromet announced the initiation of the first phase 1 clinical trial with its BiTE antibody MT110. The study will explore the safety, pharmacokinetics, pharmacodynamics and anti-tumor activity of MT110 in patients with lung cancer and patients with gastrointestinal cancer. MT110 targets the epithelial cell adhesion molecule (EpCAM or CD326), which is highly expressed on colon, lung, breast, prostate, ovarian, gastric and pancreatic cancer cells and on cancer stem cells of colon, breast, prostate and pancreas cancers. Cancer stem cells are believed to cause metastases and recurrence of these cancers.
Also in April, Micromet presented five posters at the American Association for Cancer Research (AACR) showing recent progress on the company's proprietary BiTE antibody platform and new BiTE antibodies:
-- Anti-cancer antibodies in marketed products Herceptin(R), Erbitux(R) and Vectibix(R) and the anti-asthma antibody in Xolair(R) were successfully converted to highly potent BiTE antibodies.
-- Animal data from two studies indicated feasibility of subcutaneous administration of BiTE antibodies MT103 and MT110.
-- Animal data provided proof of concept for a BiTE antibody targeting CD33 with potential use in the treatment of acute myelogenic leukemia (AML), and a BiTE antibody targeting MCSP with potential use in the treatment of melanoma.
-- Animal data suggested a therapeutic window for a BiTE antibody targeting EpCAM in a relevant animal species.
Summarizing the events, Christian Itin, Ph.D., President and Chief Executive Officer of Micromet said: "We have made significant progress with the BiTE antibody platform introducing new BiTE antibodies that target a wide range of tumor indications and converting currently marketed therapeutic antibodies into potent BiTE antibodies with much increased activity against tumor cells. We believe that the progress on our proprietary BiTE antibody platform will allow us to expand our own product pipeline, while at the same time engaging in new collaborations on selected BiTE antibody programs."
Financial Results:
Quarter Ended June 30, 2008
For the three months ended June 30, 2008, Micromet recognized total revenues of $8.5 million, compared to $3.1 million for the same period in 2007. Total operating expenses were $14.4 million for the three months ended June 30, 2008, compared to $11.1 million for the same period in 2007. For the three months ended June 30, 2008, Micromet reported a net loss of $8.6 million, or $0.21 per basic and diluted common share, compared to a net loss of $6.5 million, or $0.20 per basic and diluted common share, for the same period in 2007.
Six Months Ended June 30, 2008
For the six months ended June 30, 2008, Micromet recognized total revenues of $14.4 million, compared to $5.8 million for the same period in 2007. Total operating expenses were $27.6 million for the six months ended June 30, 2008, compared to $21.4 million for the same period in 2007. For the six months ended June 30, 2008, Micromet reported a net loss of $14.5 million, or $0.36 per basic and diluted common share, compared to a net loss of $14.1 million, or $0.44 per basic and diluted common share, for the same period in 2007.
Micromet's cash and cash equivalents were $22.4 million as of June 30, 2008. Net cash used in operating activities was $4.7 million for the six months ended June 30, 2008 compared to $7.9 million used in operating activities for the same period in 2007.
2008 Outlook:
-- At the annual meeting of the European Society for Molecular Oncology (ESMO) in September in Stockholm, Sweden, Micromet will present data from the ongoing phase 1b clinical trial of patients with metastatic breast cancer treated with adecatumumab (MT201) in combination with docetaxel. Further, Micromet plans to initiate a phase 2 clinical trial later in 2008 to evaluate adecatumumab in an adjuvant setting.
-- At the annual meeting of the American Society for Hematology (ASH) in December, Micromet expects to provide a further update on the currently ongoing phase 1 clinical trial of blinatumomab in patients with non-Hodgkins lymphoma.
-- Also at ASH, Micromet expects to present initial results on the currently ongoing phase 2 clinical trial of blinatumomab in patients with acute lymphoblastic leukemia (ALL).
-- Finally, Micromet expects that its collaboration partner MedImmune will begin a phase 1 clinical trial in the United States evaluating blinatumomab in patients with chronic lymphocytic leukemia by the end of 2008.
mfg ipollit
dies könnte für Onyx interessant sein... Bayer gilt ja als potentieller Käufer:
Presse: Bayer will weiter zukaufen
Leverkusen (aktiencheck.de AG) - Der Chemiekonzern Bayer AG (ISIN DE0005752000/ WKN 575200) geht von einer weiteren Konsolidierung innerhalb der Branche aus und fasst insbesondere im Gesundheitsgeschäft weitere Zukäufe ins Auge.
"Wir haben die Absicht, unseren Teilkonzern HealthCare weiter zu stärken. Wir wollen dort intern wie extern wachsen", erklärte Konzernchef Werner Wenning in einem Interview mit der Wirtschaftszeitung "EURO am Sonntag". Innerhalb der Kunststoffsparte MaterialScience will Wenning das Systemhausgeschäft stärken.
Im dritten Teilkonzern CropScience sieht Wenning aufgrund der starken Marktstellung bei herkömmlichen Pflanzenschutzmitteln kartellrechtlich kaum Möglichkeiten für externes Wachstum. Er sprach sich aber dafür aus, das Bioscience- und Saatgutgeschäft durch Akquisitionen auszubauen.
Die Aktie von Bayer notiert aktuell mit einem Minus von 0,78 Prozent bei 52,44 Euro. (25.08.2008/ac/n/d)
mfg ipollit
Presse: Bayer will weiter zukaufen
Leverkusen (aktiencheck.de AG) - Der Chemiekonzern Bayer AG (ISIN DE0005752000/ WKN 575200) geht von einer weiteren Konsolidierung innerhalb der Branche aus und fasst insbesondere im Gesundheitsgeschäft weitere Zukäufe ins Auge.
"Wir haben die Absicht, unseren Teilkonzern HealthCare weiter zu stärken. Wir wollen dort intern wie extern wachsen", erklärte Konzernchef Werner Wenning in einem Interview mit der Wirtschaftszeitung "EURO am Sonntag". Innerhalb der Kunststoffsparte MaterialScience will Wenning das Systemhausgeschäft stärken.
Im dritten Teilkonzern CropScience sieht Wenning aufgrund der starken Marktstellung bei herkömmlichen Pflanzenschutzmitteln kartellrechtlich kaum Möglichkeiten für externes Wachstum. Er sprach sich aber dafür aus, das Bioscience- und Saatgutgeschäft durch Akquisitionen auszubauen.
Die Aktie von Bayer notiert aktuell mit einem Minus von 0,78 Prozent bei 52,44 Euro. (25.08.2008/ac/n/d)
mfg ipollit
der Kauf von NicOx ist ein wenig unglücklich... es geht weiter deutlich nach unten. Heute hat Pfizer erklärt, dass PF-03187207 in einer PII in Asien nicht den primären Endpunkt erreicht hat wie bereits zuvor in der amerikanischen PII. Daher wird Pfizer keine PIII starten.
Zuvor ist der Kurs massiv eingebrochen, weil Goldmann Sachs deutliche Zweifel geäußert hat, ob der wichtigste Kandidat von NicOx, Naproxcinod, gegen generisches Naproxen überhaupt eine Chance besitzt und ob die Herzproblematik bei Naproxen überhaupt eine Rolle spielt. Im Erfolgsfall könnte meiner Meinung nach Naproxcinod aber nachwievor ein Blockbuster werden. Eine Ergänzung zu NicOx wäre POZN.

"NicOx' most advanced product, naproxcinod, is in phase 3 development for treating the signs and symptoms of osteoarthritis. The anti-inflammatory agents currently used in the treatment of osteoarthritis (COX-2 inhibitors and non-selective NSAIDs) have long been associated with a range of side effects, including gastrointestinal problems. Furthermore, recent clinical findings have cast significant doubt on their cardiovascular safety, including their association with increased blood pressure.
Naproxcinod is a unique nitric oxide-donating anti-inflammatory and the first of the CINOD class ("COX-Inhibiting Nitric Oxide Donators"). NicOx aims to develop naproxcinod as a powerful anti-inflammatory agent with no detrimental effects on blood pressure and good gastrointestinal tolerability and safety.
Top-line results from the first phase 3 study were successful. This represents an important step towards NicOx’ goal of making naproxcinod the drug-of-choice for osteoarthritis patients."

NicOx Says Pfizer Has Abandoned Plans To Launch Glaucoma Drug Study In Asia
8/25/2008 6:49 AM ET
(RTTNews) - Monday, French biotechnology firm NicOx S.A. (COX.PA) said that a phase II Japanese study of glaucoma drug PF-03187207 did not meet its primary end point of reduction in intraocular pressure at day 28. The company also revealed that Pfizer Inc. (PFE: News ), its partner in the development of the investigational glaucoma drug, has decided not to launch an Asian phase 3 program for PF-03187207, after reviewing the results of the Japanese study.
According to NicOx, in the phase II Japanese study, the two highest doses of PF-03187207 showed an improvement in intraocular pressure over Pfizer Inc.'s (PFE) Xalatan 0.005% of up to 11%. Abnormally raised intraocular pressure is one of the principal symptoms of glaucoma. The phase II study enrolled 112 Japanese patients with primary open-angle glaucoma or ocular hypertension.
In 2007, Xalatan raked in $1.6 billion in global sales. According to U.S. health care data provider IMS Health, sales of Xalatan in Japan totaled $250 million last year.
NiCox stated that PF-03187207 appeared to be safe and well tolerated, with only mild adverse events in the mid-stage study.
In May, the results of the U.S. study of PF-03187207, which enrolled 215 patients, showed that the highest dose of the investigational drug showed a 12% improvement over Xalatan 0.005% at day 28, but failed to attain statistical significance. Following the results of the U.S. study, Pfizer decided not to launch a phase 3 program for PF-03187207 outside of Asia.
In both the Japanese and U.S. studies, PF-03187207 showed a 20% greater reduction in intraocular pressure at 20 hours post-dose compared to Xalatan 0.005%, which reached statistical significance in the U.S. study. The results suggest that PF-03187207 has a more sustained intraocular pressure lowering effect, said NicOx.
NicOx and Pfizer are currently in discussions regarding the rights to PF-03187207, to allow its potential continued development and commercialization. Pfizer and NicOx jointly develop PF-03187207 under an August 2004 agreement. The two companies are currently concentrating on diabetic retinopathy. Diabetic retinopathy is retina damage due to diabetes, which can result in blindness.
**************
Two Ways To Play: NiCOX Gets Over Morning Bug
Terry Woo Aug 25, 2008 9:00 am
NiCOX Rebounds After Pfizer Hit
According to Bloomberg, French drug maker NicOX rebounded in European trading after its CEO said its naproxcinod, an experimental treatment for osteoarthritis, “will be a success.”
The stock rose as much as 8% reversing a decline of as much as 6% earlier in the session because Pfizer (PFE) opted not to undergo Phase III studies in Asia for its glaucoma treatment. Both companies had been working for years to develop the drug, but Pfizer decided against moving on because a study in Japan failed to reach its goal. Instead, the parties will now focus on a joint effort for a treatment of eye disease linked to diabetes.
mfg ipollit
Zuvor ist der Kurs massiv eingebrochen, weil Goldmann Sachs deutliche Zweifel geäußert hat, ob der wichtigste Kandidat von NicOx, Naproxcinod, gegen generisches Naproxen überhaupt eine Chance besitzt und ob die Herzproblematik bei Naproxen überhaupt eine Rolle spielt. Im Erfolgsfall könnte meiner Meinung nach Naproxcinod aber nachwievor ein Blockbuster werden. Eine Ergänzung zu NicOx wäre POZN.
"NicOx' most advanced product, naproxcinod, is in phase 3 development for treating the signs and symptoms of osteoarthritis. The anti-inflammatory agents currently used in the treatment of osteoarthritis (COX-2 inhibitors and non-selective NSAIDs) have long been associated with a range of side effects, including gastrointestinal problems. Furthermore, recent clinical findings have cast significant doubt on their cardiovascular safety, including their association with increased blood pressure.
Naproxcinod is a unique nitric oxide-donating anti-inflammatory and the first of the CINOD class ("COX-Inhibiting Nitric Oxide Donators"). NicOx aims to develop naproxcinod as a powerful anti-inflammatory agent with no detrimental effects on blood pressure and good gastrointestinal tolerability and safety.
Top-line results from the first phase 3 study were successful. This represents an important step towards NicOx’ goal of making naproxcinod the drug-of-choice for osteoarthritis patients."
NicOx Says Pfizer Has Abandoned Plans To Launch Glaucoma Drug Study In Asia
8/25/2008 6:49 AM ET
(RTTNews) - Monday, French biotechnology firm NicOx S.A. (COX.PA) said that a phase II Japanese study of glaucoma drug PF-03187207 did not meet its primary end point of reduction in intraocular pressure at day 28. The company also revealed that Pfizer Inc. (PFE: News ), its partner in the development of the investigational glaucoma drug, has decided not to launch an Asian phase 3 program for PF-03187207, after reviewing the results of the Japanese study.
According to NicOx, in the phase II Japanese study, the two highest doses of PF-03187207 showed an improvement in intraocular pressure over Pfizer Inc.'s (PFE) Xalatan 0.005% of up to 11%. Abnormally raised intraocular pressure is one of the principal symptoms of glaucoma. The phase II study enrolled 112 Japanese patients with primary open-angle glaucoma or ocular hypertension.
In 2007, Xalatan raked in $1.6 billion in global sales. According to U.S. health care data provider IMS Health, sales of Xalatan in Japan totaled $250 million last year.
NiCox stated that PF-03187207 appeared to be safe and well tolerated, with only mild adverse events in the mid-stage study.
In May, the results of the U.S. study of PF-03187207, which enrolled 215 patients, showed that the highest dose of the investigational drug showed a 12% improvement over Xalatan 0.005% at day 28, but failed to attain statistical significance. Following the results of the U.S. study, Pfizer decided not to launch a phase 3 program for PF-03187207 outside of Asia.
In both the Japanese and U.S. studies, PF-03187207 showed a 20% greater reduction in intraocular pressure at 20 hours post-dose compared to Xalatan 0.005%, which reached statistical significance in the U.S. study. The results suggest that PF-03187207 has a more sustained intraocular pressure lowering effect, said NicOx.
NicOx and Pfizer are currently in discussions regarding the rights to PF-03187207, to allow its potential continued development and commercialization. Pfizer and NicOx jointly develop PF-03187207 under an August 2004 agreement. The two companies are currently concentrating on diabetic retinopathy. Diabetic retinopathy is retina damage due to diabetes, which can result in blindness.
**************
Two Ways To Play: NiCOX Gets Over Morning Bug
Terry Woo Aug 25, 2008 9:00 am
NiCOX Rebounds After Pfizer Hit
According to Bloomberg, French drug maker NicOX rebounded in European trading after its CEO said its naproxcinod, an experimental treatment for osteoarthritis, “will be a success.”
The stock rose as much as 8% reversing a decline of as much as 6% earlier in the session because Pfizer (PFE) opted not to undergo Phase III studies in Asia for its glaucoma treatment. Both companies had been working for years to develop the drug, but Pfizer decided against moving on because a study in Japan failed to reach its goal. Instead, the parties will now focus on a joint effort for a treatment of eye disease linked to diabetes.
mfg ipollit
Antwort auf Beitrag Nr.: 34.854.080 von ipollit am 25.08.08 20:39:35bis jetzt war NicOx allerdings wenig erfolgreich... einige Projekte sind bereits gescheitert. Vielleicht ist es zu gewagt, auf den Erfolg von Naproxcinod zu setzen, zumal Naproxcinod von AstraZeneca verworfen wurde und NicOx bereits schon länger nach einem neuen Partner sucht, bis jetzt aber noch niemanden finden konnte. Entscheidend werden die weiteren PIII-Ergebnisse sein. Wenn es um die Magen/Darmprobleme geht, ist wohl PN-400 von POZN/AstraZeneca vielleicht aussichtsreicher...
INTERVIEW-NicOx sees growth in 2009 on new drug
Mon Jul 28, 2008 6:04pm BST
NicOx CEO expects growth in 2009 if new drug successful
* New drug Naproxcinod could lead to $1 billion annual sales
* Says all options remain open for its glaucoma drug
By Noelle Mennella
PARIS (Reuters) - 2009 could be a year of growth for NicOx (NCOX.PA: Quote, Profile, Research) if its new osteoarthritis drug Naproxcinod is approved, the French biotechnology group's Chief Executive Michele Garufi told Reuters on Monday.
In a telephone interview, Garufi said NicOx was in talks with groups in the United States to help it market the drug, which could lead to $1 billion in annual sales.
NicOx expects to file a New Drug Application (NDA) for Naproxcinod with the U.S. Food and Drug Administration (FDA) by mid-2009 when third-phase trials are completed for patients with hip and knee arthritis.
Garufi said the knee trial would be completed before the end of September and the hip trial before Christmas, adding that previous studies of the anti-inflammatory drug had shown "a very good profile" in terms of efficiency and tolerance.
"If we have a good partner, if the product's profile is confirmed, the potential is there and all the partners with whom we have spoken agree with us," Garufi said.
"Thus 2009 could be the year of transformation for NicOx. It could be the year of growth for the group in terms of business."
Garufi said NicOx, which has a market capitalisation of just under 500 million euros ($787.4 million), has had talks with several groups about marketing Naproxcinod in the United States.
"Some are in pole position. The priority is to ensure Naproxcinod's entry onto the U.S. market," he said, adding any deal would give the partner a share of the drug's sales and profits.
GLAUCOMA DRUG
Asked what would happen if Naproxcinod failed, he replied: "We have very strong finances and no debt. Clearly a failure of Naproxcinod would be very bad but it would not be the end of NicOx's technology, nor of the group."
Garufi pointed out that NicOx had also partnered with Pfizer (PFE.N: Quote, Profile, Research) in ophthalmology to develop its experimental glaucoma drug PF-03187207.
At the start of May, Pfizer, which has a 2.9 percent stake in NicOx, decided not to undertake global final Phase III tests for NicOx's drug for primary open-angle glaucoma and ocular hypertension -- diseases which can cause blindness.
However, Pfizer said it would continue the drug's development for the Asian market. A Japanese study expected at the end of September will decide on the future co-operation between the two groups.
For Garufi, "all the options are open" for PF-03187207 to be developed by Pfizer, another company, or by NicOx itself, given a sales potential of "some hundreds of millions of dollars."
On Monday the group posted a first-half loss that widened to 33.1 million euros from 6.6 million due to an increase in research spending on Naproxcinod.
NicOx shares were down 3.7 percent at 9.75 euros by 1430 GMT. The stock has shed around 10 percent in the year to date, roughly in line with the European health sector.
*********
älter von 2007!...
NicOx seeks partner for naproxcinod - looks to expand through acquisitions
PARIS, Sept 7 (APM) - NicOx is embarking on a search for a partner to market its flagship anti-inflammatory naproxcinod compound, currently in Phase III as the French biotech said it is looking to make acquisitions.
Last year, the company announced positive Phase III results for naproxcinod, a naproxene by-product, in gonarthrosis.
CEO Michele Garufi stressed that studies suggest that naproxcinod has a different profile in relation to non steroidal anti-inflammatory drugs (NSAIDs) and to COX-2 inhibitors.
He said that if the drug comes to market, it would be the "only anti inflammatory without adverse effect on arterial pressure and with good gastro intestinal tolerance", emphasising it has the potential to become a blockbuster.
Before it can submit approval requests for the product, the company needs to have the results of two other Phase III studies for gonarthrosis and osteoarthritis of the hip joint respectively.
U.S. FILING IN 2009
NicOX is looking to a U.S. filing in the first quarter of 2009 for the relief of signs and symptoms of osteoarthritis.
"Europe will be next" said Garufi, although he declined to say when a European filing is likely.
Between now and the first quarter of 2009, the company will need to find a partner for marketing naproxcinod, though he said the company is not in a hurry.
He said a 120.7 million euros capital raising move at the start of the year was a good decision.
"We did the right thing, this will allow us to continue into 2009," and giving it time to consider possible partners and ensure it secures "a good deal" for naproxcinod.
Over the next three years, the company aims to become an "integrated pharmaceutical company with commercial activities" centered on pain-related specialties and targeting cardiologists and some GPs, in the U.S. and in key European countries.
The company has a growth strategy between now and 2009, based on acquisitions or licensing deals. Garufi explained that if the company wants to increase in size, "it won't be able to live exclusively off its own products".
That strategy will be fine-tuned once the naproxcinod partnership agreement is signed, since that will determine acquisition possibilities and licensing agreements.
THREE COMPOUNDS IN PHASE II
The NicOX pipeline includes the NCX 4016 compound. It announced in June that it had deferred the start of two Phase II studies for type 2 diabetes.
This was because effects have been observed in in vitro genotoxicity pre-clinical trials on NCX 4015, a metabolite of NCX 4016.
The company has several co-development agreements - Pfizer in ophthalmology, compound PF-03187207, a prostaglandin analogue derivative that releases nitric oxide is being assessed in Phase II for glaucoma.
Topigen is carrying out Phase IIa trials on compound TPI-1020 (NCX 1020), a budesonide derivative releasing nitric oxide, in smokers who suffer from asthma. Results are due out later this year.
The company is also about to initiate a Phase IIa study on this product in chronic obstructive lung disease.
Swedish company Biolipox is running Phase II clinical trials on the compound NCX 1510 for allergic rhinitis.
Within its tie-up with Merck & Co for the development and marketing of existing class antihypertensive NO donor by-products, a first compound began Phase I in July.
Compound NCX 1047, the fruit of an agreement with the Spanish company Ferrer, is currently being pre-clinically assessed for the treatment of various dermatological pathologies.
so/cb/aki/nh
[8081] 07/09/2007 07:00 GMT - INDUSTRY
**********
NiCox launches Phase III trial of osteoarthritis drug
December 19, 2005!!!
NiCox has begun a closely watched Phase III trial for HCT-3012 for patients with osteoarthritis in the knee. HCT-3012 is derived from naproxen, and NiCox believes it could become the lead drug for osteoarthritis patients with hypertension. PRA International, a contract research organization, is managing the trial.
"We believe HCT 3012 has the potential to play a major role in the anti-inflammatory market," said Michele Garufi, chairman and CEO of NicOx. "Our view is that HCT 3012 could become the reference drug for millions of osteoarthritis patients with hypertension if this phase 3 program is successful in confirming its lack of interference with blood pressure. Initiation of this trial represents an important step toward our goal of building NicOx into a fully integrated biopharmaceutical company and we intend to retain future marketing rights to HCT-3012 in specialist areas, in line with this goal."
******************
Drugs R D. 2006 ;7 (4):262-6 16784252 (P,S,E,B) Nitronaproxen: AZD 3582, HCT 3012, Naproxen Nitroxybutylester, NO-Naproxen.
Nitronaproxen [AZD 3582, HCT 3012, naproxen nitroxybutylester, NO-naproxen] is a naproxen derivative with similar anti-inflammatory activity to the parent compound, but with less gastrointestinal toxicity. It is the first of a new class of analgesic and anti-inflammatory drugs known as cyclo-oxygenase-inhibiting nitric oxide donators (CINODs), which are under development by NicOx. The better gastrointestinal tolerability of nitronaproxen appears to be due to its release of nitric oxide (NO) and the consequent maintenance of tissue perfusion and integrity.Nitronaproxen is in phase III clinical development for the treatment of osteoarthritis and is available for licensing.AstraZeneca had been a worldwide licensee for nitronaproxen and other CINODs. However, the results of phase II clinical trials of nitronaproxen did not fulfill AstraZeneca's strategic commercial criteria for further investment and NicOx reacquired rights following AstraZeneca's decision to discontinue its involvement in 2003. NicOx was surprised by AstraZeneca's decision, and remained fully convinced of the potential of nitronaproxen. NicOx is seeking new partners for development of compounds of the CINOD class.Nitronaproxen is in a phase III clinical trial for the treatment of osteoarthritis (OA) of the knee. The 13-week trial completed enrolment of 820 patients from 120 clinical sites in the US in May 2006. The study is designed to confirm that nitronaproxen is superior to placebo and is as effective as naproxen in relieving signs and symptoms of OA. The study will also seek to show that nitronaproxen has no adverse effect on blood pressure. An additional trial has begun that is employing ambulatory blood pressure monitoring to provide a description of the blood pressure effect of nitronaproxen over a 24-hour period in hypertensive subjects. This US trial will enrol approximately 120 volunteers with stable essential hypertension. The volunteers will not have osteoarthritis but will be between the ages of 50 and 75 years (representative of the osteoarthritis population). Results from both trials are expected in the fourth quarter of 2006.The phase II clinical programme for nitronaproxen, which included 2709 patients in five separate clinical studies, showed that the drug is a potent, safe anti-inflammatory agent, with potential for improved cardiovascular safety over NSAIDs and COX-2 selective NSAIDs. An independent advisory board recommended further development of nitronaproxen in the treatment of osteoarthritis in 2004 based on an evaluation of the full results of the phase II clinical programme.A clinical study had begun in September 2004 at the University of Pennsylvania in patients with mild essential hypertension, in which the effects of nitronaproxen and rofecoxib on arterial blood pressure would be compared. However, rofecoxib was withdrawn worldwide on 1 October 2004. It is unclear if the trial was completed.The STAR Multinational Study Group has conducted a phase II gastrointestinal safety and efficacy study of nitronaproxen versus naproxen in 970 patients with osteoarthritis at 80 sites in the following countries: Argentina, Brazil, Hungary, Mexico, Norway, Poland, South Africa and the UK. The study was completed in November 2002.AstraZeneca conducted a randomised, phase II trial evaluating the efficacy and safety of nitronaproxen among 672 subjects with symptomatic knee osteoarthritis. Results have been presented.Certain phase II trial data from 2003 had been somewhat disappointing. However, an underpowered trial and failures and deficiencies in a trial meant that it was not possible to draw conclusions from this data.
******************
NicOx osteoarthritis drug fails comparison study
December 7, 2006!!!
France's NicOx says its drug naproxcinod failed to achieve its primary endpoint in a blood pressure study, but researchers touted its success for a secondary endpoint. The trial results showed a 2 mmHg difference in both the average systolic and diastolic blood pressure in favor of naproxcinod when compared to naproxen.
"We are encouraged that this study has shown a clear and positive differentiation in the blood pressure curves for naproxcinod compared to naproxen when viewed over 24 hours," said Maarten Beekman, vice president of clinical development at NicOx. "These results are in line with the difference in systolic and diastolic blood pressure that we observed for naproxcinod at two weeks in the recently announced results from the 301 phase 3 trial. We will continue to analyze the data, with a view to increasing our understanding of naproxcinod's potentially beneficial blood pressure effect."
**************
NicOx Announces Blood Pressure Analysis From 301 Phase 3 Study For Naproxcinod At EULAR
Article Date: 14 Jun 2008 - 0:00 PDT
NicOx S.A. (Euronext Paris: COX) announced an additional analysis of the blood pressure data from the 301 phase 3 study, showing a statistically significant difference between naproxcinod and naproxen in terms of the mean change from baseline in systolic and diastolic blood pressure at week 13 (p<0.05 for 3 out of 4 comparisons). Commonly used Non-Steroidal Anti-Inflammatory Drugs (NSAIDs), such as naproxen, have the tendency to raise blood pressure to an extent that may increase the rate of serious cardiovascular adverse events. The new results are being presented today at the 2008 European League Against Rheumatism (EULAR) Congress in Paris. Naproxcinod is NicOx' lead investigational drug and the first compound in the Cyclooxygenase-Inhibiting Nitric Oxide Donator (CINOD) class, which NicOx is developing for the treatment of the signs and symptoms of osteoarthritis.
Positive top-line efficacy results from the 301 study have been previously announced, showing that both doses of naproxcinod (750 mg and 375 mg bid) were superior to placebo on all three co-primary efficacy endpoints (p<0.001).Patients’ blood pressure was also measured in the study, with a well defined standardized approach whereinvestigators took office blood pressure measurements (OBPM) at each patient visit to the clinical site. The OBPM changes from baseline at week 13 were predefined safety endpoints of the study and the statistical analysis announced today was conducted on a post hoc basis.
Pascal Pfister MD, Chief Scientific Officer and Head of Research and Development at NicOx, said: “We are very pleased to be presenting these important data from our most advanced CINOD at EULAR. We believe the results of thepost hoc statistical analysis on blood pressure are particularly encouraging, as the 301 study was not powered to show a significant difference between naproxcinod and naproxen on this safety endpoint. The fact that significance has been observed in a single trial supports our confidence that a clear statistical differentiation will be obtained between naproxcinod and naproxen in the predefined pooled analysis of the OBPM data."
Two remaining phase 3 trials for naproxcinod are currently ongoing (the 302 and 303 studies) and their results are anticipated in the second half of 2008. Naproxcinod's effect on blood pressure, in comparison to naproxen and placebo, will also be assessed by OBPM in these trials, allowing NicOx to perform a predefined statistical analysis on the pooled OBPM data. NicOx projects the filing of a New Drug Application (NDA) for naproxcinod in mid-2009.
Results of the blood pressure analysis
At week 13, naproxcinod showed mean changes from baseline in systolic blood pressure, compared to naproxen, of minus 2.89 mmHg (p<0.05) for the 750 mg bid dose and -1.82 mmHg (p=0.12) for the 375 mg bid dose. For diastolic blood pressure, in terms of mean changes from baseline compared to naproxen, naproxcinod 750 mg bid was -1.79 mmHg (p<0.05) and naproxcinod 375 mg bid was -1.55 mmHg (p<0.05). At the week 13 time point, more individual patients on naproxcinod showed a decrease in blood pressure compared to patients on placebo and naproxen.
Ob sich dies mit dem Blutdruck bestätigt und dies klinisch relevant ist... NicOx sagt ja...
(sorry für die schlechte Übersetzung, aber man kann es wohl ungefähr verstehen)
(Tradingsat.com) - Après une sévère chute de l'action NicOx, toutefois en partie enrayée vendredi, Karl Hanks, porte parole de la société, fait le point sur les arguments controversés employés par le broker américain Goldman Sachs dans son étude particulièrement négative publiée la semaine dernière. (Tradingsat.com) - Nach einem schweren Sturz der Aktion NicOx, jedoch teilweise eingedämmt Freitag, Karl Hanks, Sprecher der Gesellschaft, geht auf die Argumente kontrovers Angestellten US-Broker Goldman Sachs in ihrer Studie besonders negativ in der letzten Woche veröffentlicht. L'enjeu de la discussion : le bien fondé de la stratégie visée par NicOx pour le naproxcinod, son produit phare en phase 3 de développement clinique pour le traitement des signes et symptômes de l'arthrose. Das Thema der Diskussion: die Begründetheit der Strategie nach NicOx für das naproxcinod, sein Flaggschiff-Produkt in Phase 3 der klinischen Entwicklung zur Behandlung der Anzeichen und Symptome der Arthrose.
Tradingsat.com : Goldman Sachs affirme que la littérature scientifique et médicale ne démontre pas de réelle différenciation entre le naproxcinod et le naproxene en matière d'effet sur la tension artérielle. Tradingsat.com: Goldman Sachs, dass die wissenschaftlichen und medizinischen Literatur nicht nachgewiesen, echte Differenzierung zwischen den naproxcinod und naproxene bei der Wirkung auf den Blutdruck.
Karl Hanks : Le choix des études citées par Goldman Sachs est très discutable selon nos experts. Karl Hanks: Die Auswahl der Studien von Goldman Sachs, ist sehr fraglich, nachdem wir die Experten. Dans deux d'entre elles (Hawkey, Lohmander), les patients sur lesquels était mesurée la pression artérielle faisaient l'objet d'une endoscopie. In zwei von ihnen (Hawkey, Lohmander), bei Patienten, bei denen war der Blutdruck gemessen Gegenstand einer Endoskopie. Il s'agit d'un examen plutôt angoissant qui consiste à placer une caméra dans l'estomac. Es handelt sich um eine Prüfung statt bedrohlicher, das es einer Kamera im Magen. Plusieurs études scientifiques récentes ont démontré que cet examen provoquait une augmentation du rythme cardiaque et de la pression artérielle. Mehrere wissenschaftliche Studien der jüngsten Zeit haben gezeigt, dass diese Prüfung führt zu einer Zunahme der Herzfrequenz und des Blutdrucks. Qui plus est, la façon de mesurer la pression artérielle dans ces études n'était pas standardisée de façon aussi rigoureuse que dans le programme de phase III. Wer mehr ist, wie misst man den Blutdruck in diesen Studien nicht standardisiert so streng, dass in dem Programm der Phase III.
Tradingsat.com : Comment se fait-il que Goldman Sachs affirme que la notice du naproxene ne comporte aucun avertissement sur l'hypertension, ce qui est manifestement faux ? Tradingsat.com: Wie kommt es, dass Goldman Sachs, dass die Packungsbeilage des naproxene enthält keinen Hinweis auf Bluthochdruck, was offensichtlich falsch?
Karl Hanks : Tout a fait. Karl Hanks: Alles hat. Peut-être se sont-ils basés sur une version ancienne de la notice du médicament. Möglicherweise sind sie auf der Grundlage einer älteren Version der Packungsbeilage des Arzneimittels. Il faut savoir en effet que la présence de cet avertissement relatif à la tension artérielle sur les notices de tous les anti-inflammatoires non stéroïdiens n'a été imposée par la FDA que ces dernières années. Man muss wissen, dass die Präsenz dieser Warnung über den Blutdruck über die Aufnahmen von allen nicht-steroidalen Antiphlogistika wurde durch die FDA, dass in den letzten Jahren.
Tradingsat.com : Goldman Sachs évoque également le risque d'éventuels effets secondaires liés à une libération de l'oxyde nitrique. Tradingsat.com: Goldman Sachs wird auch das Risiko möglicher Nebenwirkungen in Verbindung mit einer Freisetzung von Stickoxid.
Karl Hanks : Le risque évoqué par Goldman Sachs est purement théorique. Karl Hanks: Das Risiko, sprach von Goldman Sachs ist rein theoretisch. Comme indiqué dans notre communiqué publié le mois dernier, les résultats de l'extension sur un an de l'étude 301 n'ont révélé aucun effet secondaire anormal et confirmé le profil de sûreté et de tolérabilité du naproxcinod. Wie in unserer Pressemitteilung veröffentlicht, im vergangenen Monat die Ergebnisse der Ausweitung auf ein Jahr der Studie 301 zeigte, haben keine Nebenwirkungen und außergewöhnlichen bestätigt das Profil von Sicherheit und Verträglichkeit des naproxcinod.
Tradingsat.com : Goldman Sachs s'interroge aussi sur la dispersion des mesures sur la pression artérielle dans l'étude 301. Tradingsat.com: Goldman Sachs fragt sich auch auf die Streuung der Maßnahmen auf den Blutdruck in der Studie 301.
Karl Hanks : NicOx a récemment présenté lors du congrès EULAR (European League Against Rheumatism) » une analyse statistique sur les mesures de pression artérielle dans l'étude 301. Karl Hanks: NicOx hat vor kurzem anlässlich des EULAR-Kongress (European League Against Rheumatism) ", eine statistische Analyse über die Maßnahmen der Blutdruck in der Studie 301. Les données ont montré une différence statistiquement significative en faveur du naproxcinod, pour la pression artérielle systolique entre le naproxène 500 mg bid et le naproxcinod 750 mg bid. Die Daten zeigten einen statistisch signifikanten Unterschied zugunsten des naproxcinod, für die systolischen Blutdruck unter Naproxen 500 mg bid und naproxcinod 750 mg bid. Ce qui nous rend très confiants dans les résultats des études 302 et 303 actuellement en cours. Das macht uns sehr zuversichtlich in die Ergebnisse der Studien 302 und 303 derzeit im Gange.
Tradingsat.com : Goldman Sachs se demande par ailleurs si le traitement de certains patients avec des statines (médicaments qui réduisent le cholestérol) n'a pas pu fausser vos mesures de pression artérielles ? Tradingsat.com: Goldman Sachs fragt sich ferner, ob die Behandlung einiger Patienten mit Statinen (Medikamente, die das Cholesterin) konnte nicht verfälschen Maßnahmen zur arteriellen Druck?
Karl Hanks : C'est un fait que la population sujette à l'arthrose est souvent d'un certain âge, ce qui implique qu'elle est parfois traitée simultanément pour plusieurs pathologies telles que le cholestérol ou l'hypertension. Karl Hanks: Es ist eine Tatsache, dass die Bevölkerung abhängig von der Arthrose ist häufig von einem bestimmten Alter, was bedeutet, dass es manchmal behandelt, gleichzeitig für mehrere Krankheiten wie Bluthochdruck oder Cholesterin. Nous pouvons cependant rassurer Goldman Sachs. Wir können aber versichern, Goldman Sachs. La proportion de patients traités avec des statines dans notre panel était répartie de façon homogène entre les différents groupes de traitement de l'étude (grâce au design rigoureux de l'étude, notamment le processus de randomisation). Der Anteil der Patienten mit Statinen in unserem Gremium war gleichmäßig verteilt zwischen den verschiedenen Gruppen der Behandlung der Studie (dank der strengen Design der Studie, insbesondere den Prozess der Randomisierung).
Tradingsat.com : L'analyste semble aussi douter de la significativité d'une diminution de 2 millimètre de mercure de la tension artérielle liée à la prise de naproxcinod versus naproxène. Tradingsat.com: Der Analytiker scheint auch Zweifel an der Signifikanz einer Abnahme von 2 Millimeter Quecksilbersäule der Blutdruck im Zusammenhang mit der Einnahme von naproxcinod gegenüber Naproxen.
Karl Hanks : Selon la littérature scientifique disponible, une différence de 2 à 3 millimètres de mercure est au contraire tout à fait pertinente chez des patients traités avec des anti-inflammatoires. Karl Hanks: Nach der wissenschaftlichen Literatur verfügbar, eine Differenz von 2 bis 3 Millimeter Quecksilber ist im Gegenteil sehr relevant bei Patienten mit anti-inflammatory. Ces 2 et 3 millimètres de mercure peuvent faire une différence significative pour la mortalité et le nombre d'évènements cardiovasculaires graves. Diese 2 und 3 Millimeter Quecksilber können einen signifikanten Unterschied bei der Mortalität und die Anzahl schwerwiegender kardiovaskulärer Ereignisse.
Tradingsat.com : Sur le plan financier, Goldman Sachs estime que NicOx devra réaliser une augmentation de capital en 2009. Tradingsat.com: In finanzieller Hinsicht, Goldman Sachs ist der Ansicht, dass NicOx soll eine Kapitalerhöhung im Jahr 2009.
Karl Hanks : Ainsi que nous l'avons toujours affirmé, nous disposons de suffisamment de fonds pour financer toutes les études cliniques de phase 3 et toutes les activités jusqu'au dépôt du dossier d'enregistrement du naproxcinod auprès de la FDA mi-2009, mais aussi probablement au-delà. Karl Hanks: So, wir haben immer gesagt, wir verfügen über ausreichende Mittel zur Finanzierung aller klinischen Studien der Phase 3 und alle Aktivitäten bis zur Einreichung des Registrierungsdossiers des naproxcinod bei der FDA Mitte-2009, aber wahrscheinlich auch darüber hinaus. Il s'agissait en effet d'une prévision assez conservatrice. Es handelte sich nämlich eine recht konservative Prognose.
Propos recueillis par François Berthon Das Gespräch führte Christian Berthon
mfg ipollit
INTERVIEW-NicOx sees growth in 2009 on new drug
Mon Jul 28, 2008 6:04pm BST
NicOx CEO expects growth in 2009 if new drug successful
* New drug Naproxcinod could lead to $1 billion annual sales
* Says all options remain open for its glaucoma drug
By Noelle Mennella
PARIS (Reuters) - 2009 could be a year of growth for NicOx (NCOX.PA: Quote, Profile, Research) if its new osteoarthritis drug Naproxcinod is approved, the French biotechnology group's Chief Executive Michele Garufi told Reuters on Monday.
In a telephone interview, Garufi said NicOx was in talks with groups in the United States to help it market the drug, which could lead to $1 billion in annual sales.
NicOx expects to file a New Drug Application (NDA) for Naproxcinod with the U.S. Food and Drug Administration (FDA) by mid-2009 when third-phase trials are completed for patients with hip and knee arthritis.
Garufi said the knee trial would be completed before the end of September and the hip trial before Christmas, adding that previous studies of the anti-inflammatory drug had shown "a very good profile" in terms of efficiency and tolerance.
"If we have a good partner, if the product's profile is confirmed, the potential is there and all the partners with whom we have spoken agree with us," Garufi said.
"Thus 2009 could be the year of transformation for NicOx. It could be the year of growth for the group in terms of business."
Garufi said NicOx, which has a market capitalisation of just under 500 million euros ($787.4 million), has had talks with several groups about marketing Naproxcinod in the United States.
"Some are in pole position. The priority is to ensure Naproxcinod's entry onto the U.S. market," he said, adding any deal would give the partner a share of the drug's sales and profits.
GLAUCOMA DRUG
Asked what would happen if Naproxcinod failed, he replied: "We have very strong finances and no debt. Clearly a failure of Naproxcinod would be very bad but it would not be the end of NicOx's technology, nor of the group."
Garufi pointed out that NicOx had also partnered with Pfizer (PFE.N: Quote, Profile, Research) in ophthalmology to develop its experimental glaucoma drug PF-03187207.
At the start of May, Pfizer, which has a 2.9 percent stake in NicOx, decided not to undertake global final Phase III tests for NicOx's drug for primary open-angle glaucoma and ocular hypertension -- diseases which can cause blindness.
However, Pfizer said it would continue the drug's development for the Asian market. A Japanese study expected at the end of September will decide on the future co-operation between the two groups.
For Garufi, "all the options are open" for PF-03187207 to be developed by Pfizer, another company, or by NicOx itself, given a sales potential of "some hundreds of millions of dollars."
On Monday the group posted a first-half loss that widened to 33.1 million euros from 6.6 million due to an increase in research spending on Naproxcinod.
NicOx shares were down 3.7 percent at 9.75 euros by 1430 GMT. The stock has shed around 10 percent in the year to date, roughly in line with the European health sector.
*********
älter von 2007!...
NicOx seeks partner for naproxcinod - looks to expand through acquisitions
PARIS, Sept 7 (APM) - NicOx is embarking on a search for a partner to market its flagship anti-inflammatory naproxcinod compound, currently in Phase III as the French biotech said it is looking to make acquisitions.
Last year, the company announced positive Phase III results for naproxcinod, a naproxene by-product, in gonarthrosis.
CEO Michele Garufi stressed that studies suggest that naproxcinod has a different profile in relation to non steroidal anti-inflammatory drugs (NSAIDs) and to COX-2 inhibitors.
He said that if the drug comes to market, it would be the "only anti inflammatory without adverse effect on arterial pressure and with good gastro intestinal tolerance", emphasising it has the potential to become a blockbuster.
Before it can submit approval requests for the product, the company needs to have the results of two other Phase III studies for gonarthrosis and osteoarthritis of the hip joint respectively.
U.S. FILING IN 2009
NicOX is looking to a U.S. filing in the first quarter of 2009 for the relief of signs and symptoms of osteoarthritis.
"Europe will be next" said Garufi, although he declined to say when a European filing is likely.
Between now and the first quarter of 2009, the company will need to find a partner for marketing naproxcinod, though he said the company is not in a hurry.
He said a 120.7 million euros capital raising move at the start of the year was a good decision.
"We did the right thing, this will allow us to continue into 2009," and giving it time to consider possible partners and ensure it secures "a good deal" for naproxcinod.
Over the next three years, the company aims to become an "integrated pharmaceutical company with commercial activities" centered on pain-related specialties and targeting cardiologists and some GPs, in the U.S. and in key European countries.
The company has a growth strategy between now and 2009, based on acquisitions or licensing deals. Garufi explained that if the company wants to increase in size, "it won't be able to live exclusively off its own products".
That strategy will be fine-tuned once the naproxcinod partnership agreement is signed, since that will determine acquisition possibilities and licensing agreements.
THREE COMPOUNDS IN PHASE II
The NicOX pipeline includes the NCX 4016 compound. It announced in June that it had deferred the start of two Phase II studies for type 2 diabetes.
This was because effects have been observed in in vitro genotoxicity pre-clinical trials on NCX 4015, a metabolite of NCX 4016.
The company has several co-development agreements - Pfizer in ophthalmology, compound PF-03187207, a prostaglandin analogue derivative that releases nitric oxide is being assessed in Phase II for glaucoma.
Topigen is carrying out Phase IIa trials on compound TPI-1020 (NCX 1020), a budesonide derivative releasing nitric oxide, in smokers who suffer from asthma. Results are due out later this year.
The company is also about to initiate a Phase IIa study on this product in chronic obstructive lung disease.
Swedish company Biolipox is running Phase II clinical trials on the compound NCX 1510 for allergic rhinitis.
Within its tie-up with Merck & Co for the development and marketing of existing class antihypertensive NO donor by-products, a first compound began Phase I in July.
Compound NCX 1047, the fruit of an agreement with the Spanish company Ferrer, is currently being pre-clinically assessed for the treatment of various dermatological pathologies.
so/cb/aki/nh
[8081] 07/09/2007 07:00 GMT - INDUSTRY
**********
NiCox launches Phase III trial of osteoarthritis drug
December 19, 2005!!!
NiCox has begun a closely watched Phase III trial for HCT-3012 for patients with osteoarthritis in the knee. HCT-3012 is derived from naproxen, and NiCox believes it could become the lead drug for osteoarthritis patients with hypertension. PRA International, a contract research organization, is managing the trial.
"We believe HCT 3012 has the potential to play a major role in the anti-inflammatory market," said Michele Garufi, chairman and CEO of NicOx. "Our view is that HCT 3012 could become the reference drug for millions of osteoarthritis patients with hypertension if this phase 3 program is successful in confirming its lack of interference with blood pressure. Initiation of this trial represents an important step toward our goal of building NicOx into a fully integrated biopharmaceutical company and we intend to retain future marketing rights to HCT-3012 in specialist areas, in line with this goal."
******************
Drugs R D. 2006 ;7 (4):262-6 16784252 (P,S,E,B) Nitronaproxen: AZD 3582, HCT 3012, Naproxen Nitroxybutylester, NO-Naproxen.
Nitronaproxen [AZD 3582, HCT 3012, naproxen nitroxybutylester, NO-naproxen] is a naproxen derivative with similar anti-inflammatory activity to the parent compound, but with less gastrointestinal toxicity. It is the first of a new class of analgesic and anti-inflammatory drugs known as cyclo-oxygenase-inhibiting nitric oxide donators (CINODs), which are under development by NicOx. The better gastrointestinal tolerability of nitronaproxen appears to be due to its release of nitric oxide (NO) and the consequent maintenance of tissue perfusion and integrity.Nitronaproxen is in phase III clinical development for the treatment of osteoarthritis and is available for licensing.AstraZeneca had been a worldwide licensee for nitronaproxen and other CINODs. However, the results of phase II clinical trials of nitronaproxen did not fulfill AstraZeneca's strategic commercial criteria for further investment and NicOx reacquired rights following AstraZeneca's decision to discontinue its involvement in 2003. NicOx was surprised by AstraZeneca's decision, and remained fully convinced of the potential of nitronaproxen. NicOx is seeking new partners for development of compounds of the CINOD class.Nitronaproxen is in a phase III clinical trial for the treatment of osteoarthritis (OA) of the knee. The 13-week trial completed enrolment of 820 patients from 120 clinical sites in the US in May 2006. The study is designed to confirm that nitronaproxen is superior to placebo and is as effective as naproxen in relieving signs and symptoms of OA. The study will also seek to show that nitronaproxen has no adverse effect on blood pressure. An additional trial has begun that is employing ambulatory blood pressure monitoring to provide a description of the blood pressure effect of nitronaproxen over a 24-hour period in hypertensive subjects. This US trial will enrol approximately 120 volunteers with stable essential hypertension. The volunteers will not have osteoarthritis but will be between the ages of 50 and 75 years (representative of the osteoarthritis population). Results from both trials are expected in the fourth quarter of 2006.The phase II clinical programme for nitronaproxen, which included 2709 patients in five separate clinical studies, showed that the drug is a potent, safe anti-inflammatory agent, with potential for improved cardiovascular safety over NSAIDs and COX-2 selective NSAIDs. An independent advisory board recommended further development of nitronaproxen in the treatment of osteoarthritis in 2004 based on an evaluation of the full results of the phase II clinical programme.A clinical study had begun in September 2004 at the University of Pennsylvania in patients with mild essential hypertension, in which the effects of nitronaproxen and rofecoxib on arterial blood pressure would be compared. However, rofecoxib was withdrawn worldwide on 1 October 2004. It is unclear if the trial was completed.The STAR Multinational Study Group has conducted a phase II gastrointestinal safety and efficacy study of nitronaproxen versus naproxen in 970 patients with osteoarthritis at 80 sites in the following countries: Argentina, Brazil, Hungary, Mexico, Norway, Poland, South Africa and the UK. The study was completed in November 2002.AstraZeneca conducted a randomised, phase II trial evaluating the efficacy and safety of nitronaproxen among 672 subjects with symptomatic knee osteoarthritis. Results have been presented.Certain phase II trial data from 2003 had been somewhat disappointing. However, an underpowered trial and failures and deficiencies in a trial meant that it was not possible to draw conclusions from this data.
******************
NicOx osteoarthritis drug fails comparison study
December 7, 2006!!!
France's NicOx says its drug naproxcinod failed to achieve its primary endpoint in a blood pressure study, but researchers touted its success for a secondary endpoint. The trial results showed a 2 mmHg difference in both the average systolic and diastolic blood pressure in favor of naproxcinod when compared to naproxen.
"We are encouraged that this study has shown a clear and positive differentiation in the blood pressure curves for naproxcinod compared to naproxen when viewed over 24 hours," said Maarten Beekman, vice president of clinical development at NicOx. "These results are in line with the difference in systolic and diastolic blood pressure that we observed for naproxcinod at two weeks in the recently announced results from the 301 phase 3 trial. We will continue to analyze the data, with a view to increasing our understanding of naproxcinod's potentially beneficial blood pressure effect."
**************
NicOx Announces Blood Pressure Analysis From 301 Phase 3 Study For Naproxcinod At EULAR
Article Date: 14 Jun 2008 - 0:00 PDT
NicOx S.A. (Euronext Paris: COX) announced an additional analysis of the blood pressure data from the 301 phase 3 study, showing a statistically significant difference between naproxcinod and naproxen in terms of the mean change from baseline in systolic and diastolic blood pressure at week 13 (p<0.05 for 3 out of 4 comparisons). Commonly used Non-Steroidal Anti-Inflammatory Drugs (NSAIDs), such as naproxen, have the tendency to raise blood pressure to an extent that may increase the rate of serious cardiovascular adverse events. The new results are being presented today at the 2008 European League Against Rheumatism (EULAR) Congress in Paris. Naproxcinod is NicOx' lead investigational drug and the first compound in the Cyclooxygenase-Inhibiting Nitric Oxide Donator (CINOD) class, which NicOx is developing for the treatment of the signs and symptoms of osteoarthritis.
Positive top-line efficacy results from the 301 study have been previously announced, showing that both doses of naproxcinod (750 mg and 375 mg bid) were superior to placebo on all three co-primary efficacy endpoints (p<0.001).Patients’ blood pressure was also measured in the study, with a well defined standardized approach whereinvestigators took office blood pressure measurements (OBPM) at each patient visit to the clinical site. The OBPM changes from baseline at week 13 were predefined safety endpoints of the study and the statistical analysis announced today was conducted on a post hoc basis.
Pascal Pfister MD, Chief Scientific Officer and Head of Research and Development at NicOx, said: “We are very pleased to be presenting these important data from our most advanced CINOD at EULAR. We believe the results of thepost hoc statistical analysis on blood pressure are particularly encouraging, as the 301 study was not powered to show a significant difference between naproxcinod and naproxen on this safety endpoint. The fact that significance has been observed in a single trial supports our confidence that a clear statistical differentiation will be obtained between naproxcinod and naproxen in the predefined pooled analysis of the OBPM data."
Two remaining phase 3 trials for naproxcinod are currently ongoing (the 302 and 303 studies) and their results are anticipated in the second half of 2008. Naproxcinod's effect on blood pressure, in comparison to naproxen and placebo, will also be assessed by OBPM in these trials, allowing NicOx to perform a predefined statistical analysis on the pooled OBPM data. NicOx projects the filing of a New Drug Application (NDA) for naproxcinod in mid-2009.
Results of the blood pressure analysis
At week 13, naproxcinod showed mean changes from baseline in systolic blood pressure, compared to naproxen, of minus 2.89 mmHg (p<0.05) for the 750 mg bid dose and -1.82 mmHg (p=0.12) for the 375 mg bid dose. For diastolic blood pressure, in terms of mean changes from baseline compared to naproxen, naproxcinod 750 mg bid was -1.79 mmHg (p<0.05) and naproxcinod 375 mg bid was -1.55 mmHg (p<0.05). At the week 13 time point, more individual patients on naproxcinod showed a decrease in blood pressure compared to patients on placebo and naproxen.
Ob sich dies mit dem Blutdruck bestätigt und dies klinisch relevant ist... NicOx sagt ja...
(sorry für die schlechte Übersetzung, aber man kann es wohl ungefähr verstehen)
(Tradingsat.com) - Après une sévère chute de l'action NicOx, toutefois en partie enrayée vendredi, Karl Hanks, porte parole de la société, fait le point sur les arguments controversés employés par le broker américain Goldman Sachs dans son étude particulièrement négative publiée la semaine dernière. (Tradingsat.com) - Nach einem schweren Sturz der Aktion NicOx, jedoch teilweise eingedämmt Freitag, Karl Hanks, Sprecher der Gesellschaft, geht auf die Argumente kontrovers Angestellten US-Broker Goldman Sachs in ihrer Studie besonders negativ in der letzten Woche veröffentlicht. L'enjeu de la discussion : le bien fondé de la stratégie visée par NicOx pour le naproxcinod, son produit phare en phase 3 de développement clinique pour le traitement des signes et symptômes de l'arthrose. Das Thema der Diskussion: die Begründetheit der Strategie nach NicOx für das naproxcinod, sein Flaggschiff-Produkt in Phase 3 der klinischen Entwicklung zur Behandlung der Anzeichen und Symptome der Arthrose.
Tradingsat.com : Goldman Sachs affirme que la littérature scientifique et médicale ne démontre pas de réelle différenciation entre le naproxcinod et le naproxene en matière d'effet sur la tension artérielle. Tradingsat.com: Goldman Sachs, dass die wissenschaftlichen und medizinischen Literatur nicht nachgewiesen, echte Differenzierung zwischen den naproxcinod und naproxene bei der Wirkung auf den Blutdruck.
Karl Hanks : Le choix des études citées par Goldman Sachs est très discutable selon nos experts. Karl Hanks: Die Auswahl der Studien von Goldman Sachs, ist sehr fraglich, nachdem wir die Experten. Dans deux d'entre elles (Hawkey, Lohmander), les patients sur lesquels était mesurée la pression artérielle faisaient l'objet d'une endoscopie. In zwei von ihnen (Hawkey, Lohmander), bei Patienten, bei denen war der Blutdruck gemessen Gegenstand einer Endoskopie. Il s'agit d'un examen plutôt angoissant qui consiste à placer une caméra dans l'estomac. Es handelt sich um eine Prüfung statt bedrohlicher, das es einer Kamera im Magen. Plusieurs études scientifiques récentes ont démontré que cet examen provoquait une augmentation du rythme cardiaque et de la pression artérielle. Mehrere wissenschaftliche Studien der jüngsten Zeit haben gezeigt, dass diese Prüfung führt zu einer Zunahme der Herzfrequenz und des Blutdrucks. Qui plus est, la façon de mesurer la pression artérielle dans ces études n'était pas standardisée de façon aussi rigoureuse que dans le programme de phase III. Wer mehr ist, wie misst man den Blutdruck in diesen Studien nicht standardisiert so streng, dass in dem Programm der Phase III.
Tradingsat.com : Comment se fait-il que Goldman Sachs affirme que la notice du naproxene ne comporte aucun avertissement sur l'hypertension, ce qui est manifestement faux ? Tradingsat.com: Wie kommt es, dass Goldman Sachs, dass die Packungsbeilage des naproxene enthält keinen Hinweis auf Bluthochdruck, was offensichtlich falsch?
Karl Hanks : Tout a fait. Karl Hanks: Alles hat. Peut-être se sont-ils basés sur une version ancienne de la notice du médicament. Möglicherweise sind sie auf der Grundlage einer älteren Version der Packungsbeilage des Arzneimittels. Il faut savoir en effet que la présence de cet avertissement relatif à la tension artérielle sur les notices de tous les anti-inflammatoires non stéroïdiens n'a été imposée par la FDA que ces dernières années. Man muss wissen, dass die Präsenz dieser Warnung über den Blutdruck über die Aufnahmen von allen nicht-steroidalen Antiphlogistika wurde durch die FDA, dass in den letzten Jahren.
Tradingsat.com : Goldman Sachs évoque également le risque d'éventuels effets secondaires liés à une libération de l'oxyde nitrique. Tradingsat.com: Goldman Sachs wird auch das Risiko möglicher Nebenwirkungen in Verbindung mit einer Freisetzung von Stickoxid.
Karl Hanks : Le risque évoqué par Goldman Sachs est purement théorique. Karl Hanks: Das Risiko, sprach von Goldman Sachs ist rein theoretisch. Comme indiqué dans notre communiqué publié le mois dernier, les résultats de l'extension sur un an de l'étude 301 n'ont révélé aucun effet secondaire anormal et confirmé le profil de sûreté et de tolérabilité du naproxcinod. Wie in unserer Pressemitteilung veröffentlicht, im vergangenen Monat die Ergebnisse der Ausweitung auf ein Jahr der Studie 301 zeigte, haben keine Nebenwirkungen und außergewöhnlichen bestätigt das Profil von Sicherheit und Verträglichkeit des naproxcinod.
Tradingsat.com : Goldman Sachs s'interroge aussi sur la dispersion des mesures sur la pression artérielle dans l'étude 301. Tradingsat.com: Goldman Sachs fragt sich auch auf die Streuung der Maßnahmen auf den Blutdruck in der Studie 301.
Karl Hanks : NicOx a récemment présenté lors du congrès EULAR (European League Against Rheumatism) » une analyse statistique sur les mesures de pression artérielle dans l'étude 301. Karl Hanks: NicOx hat vor kurzem anlässlich des EULAR-Kongress (European League Against Rheumatism) ", eine statistische Analyse über die Maßnahmen der Blutdruck in der Studie 301. Les données ont montré une différence statistiquement significative en faveur du naproxcinod, pour la pression artérielle systolique entre le naproxène 500 mg bid et le naproxcinod 750 mg bid. Die Daten zeigten einen statistisch signifikanten Unterschied zugunsten des naproxcinod, für die systolischen Blutdruck unter Naproxen 500 mg bid und naproxcinod 750 mg bid. Ce qui nous rend très confiants dans les résultats des études 302 et 303 actuellement en cours. Das macht uns sehr zuversichtlich in die Ergebnisse der Studien 302 und 303 derzeit im Gange.
Tradingsat.com : Goldman Sachs se demande par ailleurs si le traitement de certains patients avec des statines (médicaments qui réduisent le cholestérol) n'a pas pu fausser vos mesures de pression artérielles ? Tradingsat.com: Goldman Sachs fragt sich ferner, ob die Behandlung einiger Patienten mit Statinen (Medikamente, die das Cholesterin) konnte nicht verfälschen Maßnahmen zur arteriellen Druck?
Karl Hanks : C'est un fait que la population sujette à l'arthrose est souvent d'un certain âge, ce qui implique qu'elle est parfois traitée simultanément pour plusieurs pathologies telles que le cholestérol ou l'hypertension. Karl Hanks: Es ist eine Tatsache, dass die Bevölkerung abhängig von der Arthrose ist häufig von einem bestimmten Alter, was bedeutet, dass es manchmal behandelt, gleichzeitig für mehrere Krankheiten wie Bluthochdruck oder Cholesterin. Nous pouvons cependant rassurer Goldman Sachs. Wir können aber versichern, Goldman Sachs. La proportion de patients traités avec des statines dans notre panel était répartie de façon homogène entre les différents groupes de traitement de l'étude (grâce au design rigoureux de l'étude, notamment le processus de randomisation). Der Anteil der Patienten mit Statinen in unserem Gremium war gleichmäßig verteilt zwischen den verschiedenen Gruppen der Behandlung der Studie (dank der strengen Design der Studie, insbesondere den Prozess der Randomisierung).
Tradingsat.com : L'analyste semble aussi douter de la significativité d'une diminution de 2 millimètre de mercure de la tension artérielle liée à la prise de naproxcinod versus naproxène. Tradingsat.com: Der Analytiker scheint auch Zweifel an der Signifikanz einer Abnahme von 2 Millimeter Quecksilbersäule der Blutdruck im Zusammenhang mit der Einnahme von naproxcinod gegenüber Naproxen.
Karl Hanks : Selon la littérature scientifique disponible, une différence de 2 à 3 millimètres de mercure est au contraire tout à fait pertinente chez des patients traités avec des anti-inflammatoires. Karl Hanks: Nach der wissenschaftlichen Literatur verfügbar, eine Differenz von 2 bis 3 Millimeter Quecksilber ist im Gegenteil sehr relevant bei Patienten mit anti-inflammatory. Ces 2 et 3 millimètres de mercure peuvent faire une différence significative pour la mortalité et le nombre d'évènements cardiovasculaires graves. Diese 2 und 3 Millimeter Quecksilber können einen signifikanten Unterschied bei der Mortalität und die Anzahl schwerwiegender kardiovaskulärer Ereignisse.
Tradingsat.com : Sur le plan financier, Goldman Sachs estime que NicOx devra réaliser une augmentation de capital en 2009. Tradingsat.com: In finanzieller Hinsicht, Goldman Sachs ist der Ansicht, dass NicOx soll eine Kapitalerhöhung im Jahr 2009.
Karl Hanks : Ainsi que nous l'avons toujours affirmé, nous disposons de suffisamment de fonds pour financer toutes les études cliniques de phase 3 et toutes les activités jusqu'au dépôt du dossier d'enregistrement du naproxcinod auprès de la FDA mi-2009, mais aussi probablement au-delà. Karl Hanks: So, wir haben immer gesagt, wir verfügen über ausreichende Mittel zur Finanzierung aller klinischen Studien der Phase 3 und alle Aktivitäten bis zur Einreichung des Registrierungsdossiers des naproxcinod bei der FDA Mitte-2009, aber wahrscheinlich auch darüber hinaus. Il s'agissait en effet d'une prévision assez conservatrice. Es handelte sich nämlich eine recht konservative Prognose.
Propos recueillis par François Berthon Das Gespräch führte Christian Berthon
mfg ipollit
Antwort auf Beitrag Nr.: 34.855.425 von ipollit am 25.08.08 22:22:29so ganz ohne scheint Naproxen nicht zu sein...
http://backandneck.about.com/od/alevenaproxen/a/aleve.htm
Intro to Aleve (naproxen)
Aleve (naproxen) is a non-steroidal anti-inflammatory (NSAID) medication used for pain relief and fever reduction. It can be purchased over the counter. Naproxen, its active ingredient, can also be obtained via prescription. Research shows that naproxen is effective in reducing the pain, tenderness, swelling and stiffness experienced by people with both inflammatory and non-inflammatory arthritis. This includes osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. People with back pain can also use Aleve for muscle aches, arthritis and other ails.
...
When Not to Take Aleve
Having certain health problems means you might not be able to take Aleve, or you may need to consult with your doctor to get the dose adjusted.
If you have a history of heart problems, blood clots, high blood pressure, stroke, kidney problems, blood clots or stomach ulcers, you should know that the class of drugs into which Aleve is categorized, known as NSAIDs, has been found to increase the risk of cardiovascular related incidents. Ask your doctor if Aleve or naproxen is right for you, and at which dosage. If it is not a good choice given your condition, perhaps your doctor can suggest a suitable substitute for pain management. The longer you take Aleve, the greater will be your risk for cardiovascular events. In particular, Aleve may increase the risk of heart attack, stroke and high blood pressure, especially with long-term use.
************
allerdings ist Naproxcinod und damit NicOx ein deutliches Risikoinvest. Sollte Naproxcinod in den laufenden PIIIs keinen signifikanten Unterschied im Blutdruck zu Naproxen erzielen, so dürfte sich NicOx vom aktuellen Kurs mehr als halbieren!
mfg ipollit
http://backandneck.about.com/od/alevenaproxen/a/aleve.htm
Intro to Aleve (naproxen)
Aleve (naproxen) is a non-steroidal anti-inflammatory (NSAID) medication used for pain relief and fever reduction. It can be purchased over the counter. Naproxen, its active ingredient, can also be obtained via prescription. Research shows that naproxen is effective in reducing the pain, tenderness, swelling and stiffness experienced by people with both inflammatory and non-inflammatory arthritis. This includes osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. People with back pain can also use Aleve for muscle aches, arthritis and other ails.
...
When Not to Take Aleve
Having certain health problems means you might not be able to take Aleve, or you may need to consult with your doctor to get the dose adjusted.
If you have a history of heart problems, blood clots, high blood pressure, stroke, kidney problems, blood clots or stomach ulcers, you should know that the class of drugs into which Aleve is categorized, known as NSAIDs, has been found to increase the risk of cardiovascular related incidents. Ask your doctor if Aleve or naproxen is right for you, and at which dosage. If it is not a good choice given your condition, perhaps your doctor can suggest a suitable substitute for pain management. The longer you take Aleve, the greater will be your risk for cardiovascular events. In particular, Aleve may increase the risk of heart attack, stroke and high blood pressure, especially with long-term use.
************
allerdings ist Naproxcinod und damit NicOx ein deutliches Risikoinvest. Sollte Naproxcinod in den laufenden PIIIs keinen signifikanten Unterschied im Blutdruck zu Naproxen erzielen, so dürfte sich NicOx vom aktuellen Kurs mehr als halbieren!
mfg ipollit
Antwort auf Beitrag Nr.: 34.639.011 von ipollit am 03.08.08 19:38:58ein gewisser Rückschlag in der Genmab-Pipeline. Amgen hat die Entwicklung von AMG-714 eingestellt!  naja, wichtiger ist es, wie Genmab mit Ofatumumab und Zalutumumab vorankommt. Aktuell beläuft sich der Cash auf ca. 440 Mio USD... bei umverändert hohem Cashburn.
naja, wichtiger ist es, wie Genmab mit Ofatumumab und Zalutumumab vorankommt. Aktuell beläuft sich der Cash auf ca. 440 Mio USD... bei umverändert hohem Cashburn.
27.08.2008 18:43
Genmab Announces 2008 First Half Year Results
COPENHAGEN, August 27 /PRNewswire-FirstCall/ -- Genmab (News) A/S (OMX: GEN) announced today results for the six month period ended June 30, 2008. During this period, Genmab reported the following results:
Genmab's revenues were DKK 277 million (USD 59 million) for the first half of 2008. In the same period of 2007, Genmab recognized revenues of DKK 280 million (USD 59 million).
An operating loss of DKK 471 million (USD 100 million). This compares to an operating loss of DKK 119 million (USD 25 million) reported for the corresponding period of 2007. The larger operating loss was driven by the increased level of pre-clinical and clinical activities associated with the advancement of our product pipeline.
Net financial income for the first half of 2008 reflected a net loss of DKK 20 million (USD 4 million), compared to a net income of DKK 32 million (USD 7 million) in the same period of 2007. The net financial income reflects a combination of interest income and fair market value adjustments from our portfolios of marketable securities and unrealized foreign exchange losses derived from the continued weakening of the USD against the DKK in the first half of 2008.
A net loss of DKK 491 million (USD 104 million) compared to a net loss of DKK 87 million (USD 18 million) for the same period in 2007. The net loss per share was DKK 11.02 (USD 2.33) for the first half of 2008 compared to DKK 2.01 (USD 0.42) in the first half of 2007.
Genmab ended the six month period with cash and marketable securities of DKK 2.1 billion (USD 443 million), which is a decrease of DKK 1.6 billion (USD 338 million) from the end of 2007. The decrease primarily arises from the DKK 1.2 billion (USD 240 million at the date of acquisition) acquisition of the manufacturing facility in March 2008.
Highlights
During the second quarter of 2008, Genmab achieved a number of business and scientific milestones, as follows:
We achieved a development milestone for ofatumumab (HuMax-CD20(R)) under the collaboration with GlaxoSmithKline (GSK). A milestone payment of DKK 29 million (USD 6 million) was triggered by the first patient participating in the Phase II study for the treatment of relapsing remitting multiple sclerosis (RRMS).
We announced a Phase I/II study to evaluate a subcutaneous route of administration of ofatumumab (HuMax-CD20(R)) in rheumatoid arthritis (RA).
We appointed David Eatwell as new Chief Financial Officer.
We initiated two Phase I/II studies of zalutumumab (HuMax-EGFr(TM)), one to treat colorectal cancer (CRC) and another study in combination with radiotherapy for the treatment of advanced head and neck cancer.
Subsequent to the balance sheet date:
We announced the positive top-line results from a Phase III pivotal study evaluating ofatumumab (HuMax-CD20(R)) in two groups of patients with chronic lymphocytic leukaemia (CLL). The study met the primary endpoint in both patient populations and the results from the secondary endpoints also supported the primary endpoint. This event also marked the achievement of a DKK 233 million (USD 49 million) milestone under the GSK collaboration agreement.
We announced plans to begin four studies of ofatumumab in CLL and NHL this year, including;
1) Phase III CLL front line chlorambucil combination study. This open-label, parallel-arm study will include 444 patients with previously untreated CLL.
2) Phase II CLL ofatumumab retreatment and maintenance treatment study. This study will examine the retreatment and maintenance treatment of refractory CLL patients who participated in the ongoing Phase III CLL study.
3) Phase II NHL ofatumumab retreatment and maintenance study. This study will examine the retreatment and maintenance treatment of refractory follicular NHL patients who participated in the ongoing Phase III NHL study.
4) Phase I study in Japan. The primary objective of the study is to evaluate the safety and tolerability of ofatumumab in Japanese relapsed/refractory follicular NHL and CLL patients.
We completed recruitment of 56 patients in the Phase II study of ofatumumab in combination with fludarabine and cyclophosphamide to treat CLL in previously untreated patients.
Amgen informed Genmab that it would discontinue development for AMG714 in psoriasis and RA based on disappointing results from recent clinical studies. Amgen is exploring options to maximize the value of this asset, but at this time no further internal development of a lead indication is planned.
Outlook
Genmab is maintaining its financial guidance for the net loss in 2008 in the range of DKK 800 to 900 million and projects a slightly improved operating loss of DKK 850 to 950 million compared to previous guidance of DKK 900 to 1,000 million. This is despite a lower revenue estimate, which is now anticipated to be in the range of DKK 850 to 900 million due to a slight change in the timing of some anticipated milestone events and lower net financial income which is now expected to be DKK 40 to 50 million. The savings are driven by a reduction in our research and development costs which is a result of our efforts to focus on the most critical programs in our portfolio in the most efficient manner. We therefore currently anticipate that we will start fewer new studies in 2008 than the 17 previously planned.
As of December 31, 2007, Genmab had cash, cash equivalents and short-term marketable securities of DKK 3.7 billion. For 2008, we project that our operations together with the DKK 1.2 billion acquisition of the manufacturing facility in Minnesota will lead to a year end cash position of DKK 1.7 to 1.8 billion (USD 359 million to USD 380 million), unchanged from previous guidance. The proportion of the budget spent on research and development, and on the ofatumumab program, also remain approximately the same as previous guidance.
mfg ipollit
 naja, wichtiger ist es, wie Genmab mit Ofatumumab und Zalutumumab vorankommt. Aktuell beläuft sich der Cash auf ca. 440 Mio USD... bei umverändert hohem Cashburn.
naja, wichtiger ist es, wie Genmab mit Ofatumumab und Zalutumumab vorankommt. Aktuell beläuft sich der Cash auf ca. 440 Mio USD... bei umverändert hohem Cashburn.27.08.2008 18:43
Genmab Announces 2008 First Half Year Results
COPENHAGEN, August 27 /PRNewswire-FirstCall/ -- Genmab (News) A/S (OMX: GEN) announced today results for the six month period ended June 30, 2008. During this period, Genmab reported the following results:
Genmab's revenues were DKK 277 million (USD 59 million) for the first half of 2008. In the same period of 2007, Genmab recognized revenues of DKK 280 million (USD 59 million).
An operating loss of DKK 471 million (USD 100 million). This compares to an operating loss of DKK 119 million (USD 25 million) reported for the corresponding period of 2007. The larger operating loss was driven by the increased level of pre-clinical and clinical activities associated with the advancement of our product pipeline.
Net financial income for the first half of 2008 reflected a net loss of DKK 20 million (USD 4 million), compared to a net income of DKK 32 million (USD 7 million) in the same period of 2007. The net financial income reflects a combination of interest income and fair market value adjustments from our portfolios of marketable securities and unrealized foreign exchange losses derived from the continued weakening of the USD against the DKK in the first half of 2008.
A net loss of DKK 491 million (USD 104 million) compared to a net loss of DKK 87 million (USD 18 million) for the same period in 2007. The net loss per share was DKK 11.02 (USD 2.33) for the first half of 2008 compared to DKK 2.01 (USD 0.42) in the first half of 2007.
Genmab ended the six month period with cash and marketable securities of DKK 2.1 billion (USD 443 million), which is a decrease of DKK 1.6 billion (USD 338 million) from the end of 2007. The decrease primarily arises from the DKK 1.2 billion (USD 240 million at the date of acquisition) acquisition of the manufacturing facility in March 2008.
Highlights
During the second quarter of 2008, Genmab achieved a number of business and scientific milestones, as follows:
We achieved a development milestone for ofatumumab (HuMax-CD20(R)) under the collaboration with GlaxoSmithKline (GSK). A milestone payment of DKK 29 million (USD 6 million) was triggered by the first patient participating in the Phase II study for the treatment of relapsing remitting multiple sclerosis (RRMS).
We announced a Phase I/II study to evaluate a subcutaneous route of administration of ofatumumab (HuMax-CD20(R)) in rheumatoid arthritis (RA).
We appointed David Eatwell as new Chief Financial Officer.
We initiated two Phase I/II studies of zalutumumab (HuMax-EGFr(TM)), one to treat colorectal cancer (CRC) and another study in combination with radiotherapy for the treatment of advanced head and neck cancer.
Subsequent to the balance sheet date:
We announced the positive top-line results from a Phase III pivotal study evaluating ofatumumab (HuMax-CD20(R)) in two groups of patients with chronic lymphocytic leukaemia (CLL). The study met the primary endpoint in both patient populations and the results from the secondary endpoints also supported the primary endpoint. This event also marked the achievement of a DKK 233 million (USD 49 million) milestone under the GSK collaboration agreement.
We announced plans to begin four studies of ofatumumab in CLL and NHL this year, including;
1) Phase III CLL front line chlorambucil combination study. This open-label, parallel-arm study will include 444 patients with previously untreated CLL.
2) Phase II CLL ofatumumab retreatment and maintenance treatment study. This study will examine the retreatment and maintenance treatment of refractory CLL patients who participated in the ongoing Phase III CLL study.
3) Phase II NHL ofatumumab retreatment and maintenance study. This study will examine the retreatment and maintenance treatment of refractory follicular NHL patients who participated in the ongoing Phase III NHL study.
4) Phase I study in Japan. The primary objective of the study is to evaluate the safety and tolerability of ofatumumab in Japanese relapsed/refractory follicular NHL and CLL patients.
We completed recruitment of 56 patients in the Phase II study of ofatumumab in combination with fludarabine and cyclophosphamide to treat CLL in previously untreated patients.
Amgen informed Genmab that it would discontinue development for AMG714 in psoriasis and RA based on disappointing results from recent clinical studies. Amgen is exploring options to maximize the value of this asset, but at this time no further internal development of a lead indication is planned.
Outlook
Genmab is maintaining its financial guidance for the net loss in 2008 in the range of DKK 800 to 900 million and projects a slightly improved operating loss of DKK 850 to 950 million compared to previous guidance of DKK 900 to 1,000 million. This is despite a lower revenue estimate, which is now anticipated to be in the range of DKK 850 to 900 million due to a slight change in the timing of some anticipated milestone events and lower net financial income which is now expected to be DKK 40 to 50 million. The savings are driven by a reduction in our research and development costs which is a result of our efforts to focus on the most critical programs in our portfolio in the most efficient manner. We therefore currently anticipate that we will start fewer new studies in 2008 than the 17 previously planned.
As of December 31, 2007, Genmab had cash, cash equivalents and short-term marketable securities of DKK 3.7 billion. For 2008, we project that our operations together with the DKK 1.2 billion acquisition of the manufacturing facility in Minnesota will lead to a year end cash position of DKK 1.7 to 1.8 billion (USD 359 million to USD 380 million), unchanged from previous guidance. The proportion of the budget spent on research and development, and on the ofatumumab program, also remain approximately the same as previous guidance.
mfg ipollit
zu Intercell...
Donnerstag, 28. August 2008, 09:17
S. aureus: Weitere Phase-II-Studie mit V710
Intercell gab bekannt, dass ihr Partner Merck & Co eine Phase-II-Studie mit dem Impfstoffkandidaten V710 – er basiert auf einem von Intercell entdeckten hochkonservierten Protein-Antigen – zur Vorbeugung von S. aureus Infektionen gestartet hat.
In dieser randomisierten, placebokontrollierten Doppelblind-Studie soll die Sicherheit und Wirksamkeit des Impfstoffs bei Hämodialyse-Patienten untersucht werden. Die Studie erweitert die im Dezember gestartete Phase-II-Studie.
Das von Intercell entdeckte Antigen wurde 2004 mit Merck & Co in eine weltweite exklusive Lizenzpartnerschaft eingebracht. Merck hat hierbei die Verantwortung für die klinische Entwicklung, die Herstellung und das Marketing übernommen. Intercell erhält Meilensteinzahlungen und Lizenzgebühren auf künftige Verkaufserlöse.
Die bereits abgeschlossenen Phase-I-Studien zeigten, dass der Impfstoffkandidat gegen S. aureus immunogen, sicher und allgemein sehr gut verträglich ist.
Parallel dazu arbeitet Intercell an einem Impfstoff, der gegen im Krankenhaus erworbene Pseudomonas aeruginosa Infektionen eingesetzt werden soll – eine Phase-II/III-Studie ist für 2008 geplant. Darüber hinaus befindet sich ein Impfstoffkandidat gegen Pneumokokken in der Pipeline von Intercell (Start von Phase-I-Studien ebenso noch heuer geplant). Ebenso werden präklinische Programme zur Entwicklung von Impfstoffen gegen Enterococcus und Klebsiella – Keime, die ebenfalls zu Infektionen im Krankenhaus führen – durchgeführt.
mfg ipollit
Donnerstag, 28. August 2008, 09:17
S. aureus: Weitere Phase-II-Studie mit V710
Intercell gab bekannt, dass ihr Partner Merck & Co eine Phase-II-Studie mit dem Impfstoffkandidaten V710 – er basiert auf einem von Intercell entdeckten hochkonservierten Protein-Antigen – zur Vorbeugung von S. aureus Infektionen gestartet hat.
In dieser randomisierten, placebokontrollierten Doppelblind-Studie soll die Sicherheit und Wirksamkeit des Impfstoffs bei Hämodialyse-Patienten untersucht werden. Die Studie erweitert die im Dezember gestartete Phase-II-Studie.
Das von Intercell entdeckte Antigen wurde 2004 mit Merck & Co in eine weltweite exklusive Lizenzpartnerschaft eingebracht. Merck hat hierbei die Verantwortung für die klinische Entwicklung, die Herstellung und das Marketing übernommen. Intercell erhält Meilensteinzahlungen und Lizenzgebühren auf künftige Verkaufserlöse.
Die bereits abgeschlossenen Phase-I-Studien zeigten, dass der Impfstoffkandidat gegen S. aureus immunogen, sicher und allgemein sehr gut verträglich ist.
Parallel dazu arbeitet Intercell an einem Impfstoff, der gegen im Krankenhaus erworbene Pseudomonas aeruginosa Infektionen eingesetzt werden soll – eine Phase-II/III-Studie ist für 2008 geplant. Darüber hinaus befindet sich ein Impfstoffkandidat gegen Pneumokokken in der Pipeline von Intercell (Start von Phase-I-Studien ebenso noch heuer geplant). Ebenso werden präklinische Programme zur Entwicklung von Impfstoffen gegen Enterococcus und Klebsiella – Keime, die ebenfalls zu Infektionen im Krankenhaus führen – durchgeführt.
mfg ipollit
Zur Genmab Webcast.
Habt ihr euch heute den webcast von Genmab angehört ?
Es gab eine sehr wichtige Aussage zur Fase 3 Versuch von
Humax-Egfr . Es wurde gesagt 'Die Patienten überleben länger'. Deshalb kommen die Resultate nicht in Q4 sondern erst feb/mar 2009 !
Dass könnte darauf hindeuten, dass die patienten mit Egfr behandlung mindestens dobbelt so lange überleben als Erbitux beh. Patienten.
Habt ihr euch heute den webcast von Genmab angehört ?
Es gab eine sehr wichtige Aussage zur Fase 3 Versuch von
Humax-Egfr . Es wurde gesagt 'Die Patienten überleben länger'. Deshalb kommen die Resultate nicht in Q4 sondern erst feb/mar 2009 !
Dass könnte darauf hindeuten, dass die patienten mit Egfr behandlung mindestens dobbelt so lange überleben als Erbitux beh. Patienten.
Zur Genmab Webcast.
Habt ihr euch heute den webcast von Genmab angehört ?
Es gab eine sehr wichtige Aussage zur Fase 3 Versuch von
Humax-Egfr . Es wurde gesagt 'Die Patienten überleben länger'. Deshalb kommen die Resultate nicht in Q4 sondern erst feb/mar 2009 !
Dass könnte darauf hindeuten, dass die patienten mit Egfr behandlung mindestens dobbelt so lange überleben als Erbitux beh. Patienten.
Habt ihr euch heute den webcast von Genmab angehört ?
Es gab eine sehr wichtige Aussage zur Fase 3 Versuch von
Humax-Egfr . Es wurde gesagt 'Die Patienten überleben länger'. Deshalb kommen die Resultate nicht in Q4 sondern erst feb/mar 2009 !
Dass könnte darauf hindeuten, dass die patienten mit Egfr behandlung mindestens dobbelt so lange überleben als Erbitux beh. Patienten.
die Charts des aktuellen Depots...
Genmab

ONXX
Regeneron
Evotec
Cubist
Intercell
Isis
Exelixis

Micromet
Array

Arena
Addex

ViroPharma
OSI Pharma
Incyte

Rigel

Sucampo

NicOx
mfg ipollit
Genmab
ONXX
Regeneron
Evotec
Cubist
Intercell
Isis
Exelixis
Micromet
Array
Arena
Addex
ViroPharma
OSI Pharma
Incyte
Rigel
Sucampo
NicOx
mfg ipollit
Antwort auf Beitrag Nr.: 34.900.182 von ipollit am 28.08.08 23:50:00zum Vergleich der Nasdaq-Biotech Index
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 34.900.071 von steelbooming am 28.08.08 23:34:47Danke für die Info... werde ich noch machen. Ich bin allerdings vorsichtig mit irgendwelchen Rückschlüssen... warte lieber ab, was die Ergebnisse sagen. Dieses "die Patienten leben länger" war schon bei GPC ein Trugschluss.
Allerdings sehe ich HuMax-EGFR als sehr interessant, speziell auch in Hinblick auf die aktuelle IMCL-Übernahme.
mfg ipollit
Allerdings sehe ich HuMax-EGFR als sehr interessant, speziell auch in Hinblick auf die aktuelle IMCL-Übernahme.
mfg ipollit
Antwort auf Beitrag Nr.: 34.900.182 von ipollit am 28.08.08 23:50:00Watch-Liste... interessant finde ich aktuell Progenix. Außerdem habe ich mir NeuroSearch angesehen... ist auch nicht schlecht.
Progenix

Pozen

Curis

Momenta

Ariad

Trubion

Medigene

Cytos

NeuroSearch

mfg ipollit
Progenix
Pozen
Curis
Momenta
Ariad
Trubion
Medigene
Cytos
NeuroSearch
mfg ipollit
Änderung seit Montag...
- Position in Progenics
- Regeneron verringert
Genmab 21,7% http://finance.yahoo.com/q?s=GEN.CO
Onyx 9,0% http://finance.yahoo.com/q?s=ONXX
Evotec 8,4% http://finance.yahoo.com/q?s=EVT.DE
Regeneron 7,0% http://finance.yahoo.com/q?s=REGN
Cubist 5,1% http://finance.yahoo.com/q?s=CBST
Intercell 5,1% http://finance.yahoo.com/q?s=IJE.F
Isis 4,9% http://finance.yahoo.com/q?s=ISIS
Exelixis 4,5% http://finance.yahoo.com/q?s=EXEL
Array 4,0% http://finance.yahoo.com/q?s=ARRY
Micromet 3,9% http://finance.yahoo.com/q?s=MITI
Progenics 3,8% http://finance.yahoo.com/q?s=PGNX
ViroPharma 3,5% http://finance.yahoo.com/q?s=VPHM
Addex 3,5% http://finance.yahoo.com/q?s=ADXN.SW
Arena 3,5% http://finance.yahoo.com/q?s=ARNA
OSI 2,9% http://finance.yahoo.com/q?s=OSIP
Rigel 2,6% http://finance.yahoo.com/q?s=RIGL
Incyte 2,5% http://finance.yahoo.com/q?s=INCY
Sucampo 2,2% http://finance.yahoo.com/q?s=SCMP
NicOx 2,0% http://finance.yahoo.com/q?s=COX.PA
mfg ipollit
- Position in Progenics
- Regeneron verringert
Genmab 21,7% http://finance.yahoo.com/q?s=GEN.CO
Onyx 9,0% http://finance.yahoo.com/q?s=ONXX
Evotec 8,4% http://finance.yahoo.com/q?s=EVT.DE
Regeneron 7,0% http://finance.yahoo.com/q?s=REGN
Cubist 5,1% http://finance.yahoo.com/q?s=CBST
Intercell 5,1% http://finance.yahoo.com/q?s=IJE.F
Isis 4,9% http://finance.yahoo.com/q?s=ISIS
Exelixis 4,5% http://finance.yahoo.com/q?s=EXEL
Array 4,0% http://finance.yahoo.com/q?s=ARRY
Micromet 3,9% http://finance.yahoo.com/q?s=MITI
Progenics 3,8% http://finance.yahoo.com/q?s=PGNX
ViroPharma 3,5% http://finance.yahoo.com/q?s=VPHM
Addex 3,5% http://finance.yahoo.com/q?s=ADXN.SW
Arena 3,5% http://finance.yahoo.com/q?s=ARNA
OSI 2,9% http://finance.yahoo.com/q?s=OSIP
Rigel 2,6% http://finance.yahoo.com/q?s=RIGL
Incyte 2,5% http://finance.yahoo.com/q?s=INCY
Sucampo 2,2% http://finance.yahoo.com/q?s=SCMP
NicOx 2,0% http://finance.yahoo.com/q?s=COX.PA
mfg ipollit
zu Progenics...
GSK ist bei Adolor ausgestiegen, was die interessanteste Indikation chronische OBD betrifft. Damit ist der Weg für Progenics/Wyeth frei... interessant wird auch sein, wie Relistor nun am Markt angekommen ist.

Tuesday, September 2, 2008 - 10:25 AM EDT
GlaxoSmithKline scales back relationship with AdolorPittsburgh Business Times
Print Email Reprints RSS Feeds Add to Del.icio.us Digg This CommentsGlaxoSmithKline has returned to Adolor Corp. the worldwide rights related to the development of the Entereg drug for chronic opioid bowel dysfunction.
GlaxoSmithKline is retaining rights to Entereg for postoperative ileus (POI), an impairment of gastrointestinal function following abdominal surgery.
Adolor (NASDAQ:ADLR) officials said Tuesday they plan to pursue usage of Entereg for chronic opioid bowel dysfunction.
“There is a large, unmet need for treatment options for the many patients who suffer with chronic OBD,” said Michael R. Dougherty, president and CEO of the Exton, Pa., biopharmaceutical company. “Adolor maintains a portfolio of development candidates that may potentially serve this patient population, including Entereg, our combination product program and additional earlier stage compounds. We intend now to explore discussions with potential partners regarding this portfolio, and to submit to the (Food and Drug Administration) for review a protocol for an additional study of Entereg in OBD under a special protocol assessment.”
Opioid bowel dysfunction occurs both with short- and long-term use of opioid pain medicines, and it is characterized by constipation, bloating and other bowel dysfunction.
GSK (NYSE:GSK) gave no reason for returning the product rights, and information on the financial impact of the company’s move was not provided.
Entereg was approved by the FDA in May for postoperative ileus. The companies will continue to collaborate on the development and commercialization of Entereg for POI in the United States.
Based in London, GlaxoSmithKline has a U.S. headquarters in Philadelphia and a consumer health care division near Pittsburgh.
mfg ipollit
GSK ist bei Adolor ausgestiegen, was die interessanteste Indikation chronische OBD betrifft. Damit ist der Weg für Progenics/Wyeth frei... interessant wird auch sein, wie Relistor nun am Markt angekommen ist.
Tuesday, September 2, 2008 - 10:25 AM EDT
GlaxoSmithKline scales back relationship with AdolorPittsburgh Business Times
Print Email Reprints RSS Feeds Add to Del.icio.us Digg This CommentsGlaxoSmithKline has returned to Adolor Corp. the worldwide rights related to the development of the Entereg drug for chronic opioid bowel dysfunction.
GlaxoSmithKline is retaining rights to Entereg for postoperative ileus (POI), an impairment of gastrointestinal function following abdominal surgery.
Adolor (NASDAQ:ADLR) officials said Tuesday they plan to pursue usage of Entereg for chronic opioid bowel dysfunction.
“There is a large, unmet need for treatment options for the many patients who suffer with chronic OBD,” said Michael R. Dougherty, president and CEO of the Exton, Pa., biopharmaceutical company. “Adolor maintains a portfolio of development candidates that may potentially serve this patient population, including Entereg, our combination product program and additional earlier stage compounds. We intend now to explore discussions with potential partners regarding this portfolio, and to submit to the (Food and Drug Administration) for review a protocol for an additional study of Entereg in OBD under a special protocol assessment.”
Opioid bowel dysfunction occurs both with short- and long-term use of opioid pain medicines, and it is characterized by constipation, bloating and other bowel dysfunction.
GSK (NYSE:GSK) gave no reason for returning the product rights, and information on the financial impact of the company’s move was not provided.
Entereg was approved by the FDA in May for postoperative ileus. The companies will continue to collaborate on the development and commercialization of Entereg for POI in the United States.
Based in London, GlaxoSmithKline has a U.S. headquarters in Philadelphia and a consumer health care division near Pittsburgh.
mfg ipollit
Antwort auf Beitrag Nr.: 34.970.784 von ipollit am 03.09.08 23:47:36"Änderung seit Montag..." genau genommen gestern.
zu Regeneron...
die Indikationserweiterung von Arcalyst auf die weit größere Indikation Gicht sieht gut aus... die PII war positiv. Noch in diesem Jahr soll die PIII gestartet werden.
http://www.nytimes.com/2008/09/03/health/research/03gout.htm…
A Regeneron Drug Minimized Recurrence of Gout in Clinical Trial
By ANDREW POLLACK
Published: September 2, 2008
A drug being developed by Regeneron Pharmaceuticals sharply reduced the number of gout flare-ups in a clinical trial, the company is expected to announce Wednesday.
Experts said the drug, called Arcalyst, potentially could be a significant advance in treatment of gout, an intensely painful joint inflammation that, according to the American College of Rheumatology, afflicts up to three million Americans.
“You can really dramatically blunt the risk of having flares,” said Dr. H. Ralph Schumacher Jr., a professor of medicine at the University of Pennsylvania who is a consultant to Regeneron.
The results, though, were from a midstage trial involving only 83 patients who were monitored for just 12 weeks. Regeneron’s chief executive, Leonard Schleifer, said the company planned to begin larger, Phase 3 studies — the kind required to seek federal approval of a drug — by early next year.
Moreover, Arcalyst was tested against a placebo, not against other drugs now used to reduce the risk of gout flare-ups. Those treatments are inexpensive pills like naproxen, while Arcalyst must be injected once a week and is likely to be far more expensive.
Arcalyst, also known as rilonacept or IL-1 Trap, was approved in February as a treatment for a group of extremely rare inflammatory diseases called cryopirin-associated periodic syndromes, which can cause fevers, chills and joint pain. Each weekly injection costs about $5,000, so it would be prohibitively expensive for doctors to use Arcalyst off-label to treat gout. The company is expecting sales of about $10 million this year for the medicine, which is its first drug to reach the market.
Regeneron, which is based in Tarrytown, N.Y., has said it would sharply reduce the price if the drug were to win approval as a treatment for a more common condition like gout.
Gout occurs when uric acid builds up in the blood to the point where it can no longer stay in solution. It forms crystals that lodge in the joints, often of the big toe.
Patients can take drugs, particularly one called allopurinol, to reduce the level of uric acid and eventually eliminate symptoms of gout. But for reasons not fully understood, the effect of such treatment over the first several months is to increase the risk of gout’s flaring up.
“People may actually sense they are getting worse before they are getting better, and to some extent it’s true,” said Dr. John S. Sundy, an associate professor at Duke who has gotten research support and consulting fees from Regeneron. That discourages many patients from starting treatment to reduce uric acid levels.
The Arcalyst clinical trial was intended to see if the drug could prevent flare-ups, which can last up to 10 days, in patients starting treatment with allopurinol.
Only 14.6 percent of the patients who received Arcalyst experienced a flare-up over the 12 weeks of the trial, and none had more than one flare-up. Among those treated with a placebo, 45.2 percent had at least one flare-up, and many of them had more than one.
The mean number of flare-ups per patient was 0.15 with Arcalyst and 0.79 with placebo, an 81 percent reduction.
Arcalyst works by blocking the action of an inflammatory protein called interleukin-1.
mfg ipollit
die Indikationserweiterung von Arcalyst auf die weit größere Indikation Gicht sieht gut aus... die PII war positiv. Noch in diesem Jahr soll die PIII gestartet werden.
http://www.nytimes.com/2008/09/03/health/research/03gout.htm…
A Regeneron Drug Minimized Recurrence of Gout in Clinical Trial
By ANDREW POLLACK
Published: September 2, 2008
A drug being developed by Regeneron Pharmaceuticals sharply reduced the number of gout flare-ups in a clinical trial, the company is expected to announce Wednesday.
Experts said the drug, called Arcalyst, potentially could be a significant advance in treatment of gout, an intensely painful joint inflammation that, according to the American College of Rheumatology, afflicts up to three million Americans.
“You can really dramatically blunt the risk of having flares,” said Dr. H. Ralph Schumacher Jr., a professor of medicine at the University of Pennsylvania who is a consultant to Regeneron.
The results, though, were from a midstage trial involving only 83 patients who were monitored for just 12 weeks. Regeneron’s chief executive, Leonard Schleifer, said the company planned to begin larger, Phase 3 studies — the kind required to seek federal approval of a drug — by early next year.
Moreover, Arcalyst was tested against a placebo, not against other drugs now used to reduce the risk of gout flare-ups. Those treatments are inexpensive pills like naproxen, while Arcalyst must be injected once a week and is likely to be far more expensive.
Arcalyst, also known as rilonacept or IL-1 Trap, was approved in February as a treatment for a group of extremely rare inflammatory diseases called cryopirin-associated periodic syndromes, which can cause fevers, chills and joint pain. Each weekly injection costs about $5,000, so it would be prohibitively expensive for doctors to use Arcalyst off-label to treat gout. The company is expecting sales of about $10 million this year for the medicine, which is its first drug to reach the market.
Regeneron, which is based in Tarrytown, N.Y., has said it would sharply reduce the price if the drug were to win approval as a treatment for a more common condition like gout.
Gout occurs when uric acid builds up in the blood to the point where it can no longer stay in solution. It forms crystals that lodge in the joints, often of the big toe.
Patients can take drugs, particularly one called allopurinol, to reduce the level of uric acid and eventually eliminate symptoms of gout. But for reasons not fully understood, the effect of such treatment over the first several months is to increase the risk of gout’s flaring up.
“People may actually sense they are getting worse before they are getting better, and to some extent it’s true,” said Dr. John S. Sundy, an associate professor at Duke who has gotten research support and consulting fees from Regeneron. That discourages many patients from starting treatment to reduce uric acid levels.
The Arcalyst clinical trial was intended to see if the drug could prevent flare-ups, which can last up to 10 days, in patients starting treatment with allopurinol.
Only 14.6 percent of the patients who received Arcalyst experienced a flare-up over the 12 weeks of the trial, and none had more than one flare-up. Among those treated with a placebo, 45.2 percent had at least one flare-up, and many of them had more than one.
The mean number of flare-ups per patient was 0.15 with Arcalyst and 0.79 with placebo, an 81 percent reduction.
Arcalyst works by blocking the action of an inflammatory protein called interleukin-1.
mfg ipollit
ist auch nicht uninteressant: ZGEN
mit Recothrom ist bereits ein Produkt am Markt. Der heutige Deal mit Merck reduziert weiter die Kosten und Risiken.

The companies also changed their research deal, under which ZymoGenetics will now control the development and commercialization of IL-31 mAb, and Merck Serono will manage the same for IL-17RC.
Both compounds are being tested as treatments for inflammatory diseases and are currently in preclinical stages. (Reporting by Varsha Tickoo in Bangalore; Editing by Himani Sarkar)
mfg ipollit" target="_blank" rel="nofollow ugc noopener">Tuesday, September 2, 2008 - 9:38 AM PDT
ZymoGenetics gets …[/i]
mit Recothrom ist bereits ein Produkt am Markt. Der heutige Deal mit Merck reduziert weiter die Kosten und Risiken.
The companies also changed their research deal, under which ZymoGenetics will now control the development and commercialization of IL-31 mAb, and Merck Serono will manage the same for IL-17RC.
Both compounds are being tested as treatments for inflammatory diseases and are currently in preclinical stages. (Reporting by Varsha Tickoo in Bangalore; Editing by Himani Sarkar)
mfg ipollit" target="_blank" rel="nofollow ugc noopener">Tuesday, September 2, 2008 - 9:38 AM PDT
ZymoGenetics gets …[/i]
Antwort auf Beitrag Nr.: 34.900.045 von steelbooming am 28.08.08 23:31:38hier ist auch noch eine Aussage zu Genmab's möglichen Erbitux-Konkurrenten Zalutumumab und den verzögerten PIII-Ergebnissen. Sollten die Ergebnisse, die jetzt eher im März nächsten Jahres erwartet werden, positiv sein, stehen die Chancen laut Genmab auf einen 1+ Mrd USD Deal gut.
http://www.reuters.com/article/companyNews/idUKL423615420080…
Genmab CEO eyes next big drug deal in 2009
Fri Sep 5, 2008 2:44am EDT
By Ben Hirschler
LONDON (Reuters) - Danish biotech company Genmab (GEN.CO: Quote, Profile, Research, Stock Buzz) could sign a $1 billion-plus (569,000 pounds-plus) partnership deal for its experimental cancer drug zalutumumab next year if interim clinical trial results, expected in March, are positive.
Chief Executive Lisa Drakeman said on Thursday that partnering the product -- a rival to ImClone's (IMCL.O: Quote, Profile, Research, Stock Buzz) Erbitux -- was the logical next step.
"If the interim data is significant, then I would say we would be in a position to immediately start really serious discussions," she told Reuters, during a visit to London.
"But it would probably realistically take up to six months, though it could take three or four months, to sign a partnership."
Genmab said last week that survival data with zalutumumab, also known as HuMax-EGFr, were not expected to be available until March 2009. They had been expected by the end of the year.
The delay suggests that patients on the drug in the Phase III trial may be living longer, since patients are randomised 2:1 to get either zalutumumab or best supportive care.
"The chances are pretty good, actually, that we are having better survival in the treatment arm," Drakeman said.
Zalutumumab is being tested initially as a treatment for head and neck cancer but earlier-stage trials are also in progress against lung and bowel cancer.
Sales of Erbitux, the leading so-called EGFr cancer treatment, are now more than $1 billion a year. The drug is sold by Merck KGaA (MRCG.DE: Quote, Profile, Research, Stock Buzz) in Europe and by Bristol-Myers Squibb (BMY.N: Quote, Profile, Research, Stock Buzz) in the United States.
Drakeman said the commercial opportunity for cancer drugs targeting the EGFr protein were smaller than those against CD20, for which the leading product is Roche (ROG.VX: Quote, Profile, Research, Stock Buzz) and Genentech's (DNA.N: Quote, Profile, Research, Stock Buzz) Rituxan/MabThera.
Genmab signed a record deal worth up $2.1 billion with GlaxoSmithKline (GSK.L: Quote, Profile, Research, Stock Buzz) in December 2006 for its leading CD20 antibody ofatumumab, which may reach the market in 2009.
"Realistically, we shouldn't expect as big a transaction for this antibody. On the other hand, if it looks great on the Phase III data, we'd be at a different point than we were with the CD20 antibody, where we'd just started Phase III," Drakeman said.
Asked if a zalutumumab deal was likely to be worth more than $1 billion, she said: "That seems reasonable."
mfg ipollit
http://www.reuters.com/article/companyNews/idUKL423615420080…
Genmab CEO eyes next big drug deal in 2009
Fri Sep 5, 2008 2:44am EDT
By Ben Hirschler
LONDON (Reuters) - Danish biotech company Genmab (GEN.CO: Quote, Profile, Research, Stock Buzz) could sign a $1 billion-plus (569,000 pounds-plus) partnership deal for its experimental cancer drug zalutumumab next year if interim clinical trial results, expected in March, are positive.
Chief Executive Lisa Drakeman said on Thursday that partnering the product -- a rival to ImClone's (IMCL.O: Quote, Profile, Research, Stock Buzz) Erbitux -- was the logical next step.
"If the interim data is significant, then I would say we would be in a position to immediately start really serious discussions," she told Reuters, during a visit to London.
"But it would probably realistically take up to six months, though it could take three or four months, to sign a partnership."
Genmab said last week that survival data with zalutumumab, also known as HuMax-EGFr, were not expected to be available until March 2009. They had been expected by the end of the year.
The delay suggests that patients on the drug in the Phase III trial may be living longer, since patients are randomised 2:1 to get either zalutumumab or best supportive care.
"The chances are pretty good, actually, that we are having better survival in the treatment arm," Drakeman said.
Zalutumumab is being tested initially as a treatment for head and neck cancer but earlier-stage trials are also in progress against lung and bowel cancer.
Sales of Erbitux, the leading so-called EGFr cancer treatment, are now more than $1 billion a year. The drug is sold by Merck KGaA (MRCG.DE: Quote, Profile, Research, Stock Buzz) in Europe and by Bristol-Myers Squibb (BMY.N: Quote, Profile, Research, Stock Buzz) in the United States.
Drakeman said the commercial opportunity for cancer drugs targeting the EGFr protein were smaller than those against CD20, for which the leading product is Roche (ROG.VX: Quote, Profile, Research, Stock Buzz) and Genentech's (DNA.N: Quote, Profile, Research, Stock Buzz) Rituxan/MabThera.
Genmab signed a record deal worth up $2.1 billion with GlaxoSmithKline (GSK.L: Quote, Profile, Research, Stock Buzz) in December 2006 for its leading CD20 antibody ofatumumab, which may reach the market in 2009.
"Realistically, we shouldn't expect as big a transaction for this antibody. On the other hand, if it looks great on the Phase III data, we'd be at a different point than we were with the CD20 antibody, where we'd just started Phase III," Drakeman said.
Asked if a zalutumumab deal was likely to be worth more than $1 billion, she said: "That seems reasonable."
mfg ipollit
Antwort auf Beitrag Nr.: 34.970.851 von ipollit am 03.09.08 23:55:29noch zu Progenics vs. Adolor...
http://www.tmcnet.com/usubmit/2008/09/02/3629494.htm
September 02, 2008
GSK Hands Entereg OBD Indication Back to Adolor
(BioWorld Today Via Acquire Media NewsEdge) Drug giant GlaxoSmithKline Plc handed back rights to bowel drug Entereg to Adolor Corp., a setback for GSK's partner but good news for Progenics Pharmaceuticals Inc., which has its own bowel drug Relistor that could compete with Entereg. Both Adolor and Progenics are seeking to expand use of their respective FDA-approved bowel drugs.
But Adolor's plans could be delayed as a result of GSK walking away from a program evaluating Entereg in chronic opioid-induced bowel dysfunction. Meantime, Progenics could move out front, with a Phase III study of its bowel drug expected next year.
"Anything that slows down Entereg will be good for Relistor," Need ham & Co. analyst Alan Carr told BioWorld Today. Carr, who tracks Progenics, said the setback for Adolor's Entereg gives Progenics "more breathing room."
Adolor will regain the rights to Entereg in opioid bowel dysfunction, or OBD, and will likely need to find another partner to advance Entereg in that indication. Now that the FDA has lifted a clinical hold on Entereg for OBD, Adolor is considering conducting another study under a special protocol assessment.
Michael R. Dougherty, president and CEO of Adolor, said in a statement that "Adolor maintains a portfolio of development candidates that may potentially serve this patient population, including Entereg, our combination product program, and additional earlier stage compounds. We intend now to explore discussions with potential partners regarding this portfolio, and to submit to the FDA for review a protocol for an additional study of Entereg in OBD under a special protocol assessment."
Adolor's Entereg currently is approved for postoperative ileus, in which the bowels lose their motility. GSK is retaining rights to Entereg for postoperative ileus (POI), and the companies will continue to collaborate on the development and commercialization of Entereg for POI in the US., Adolor said.
The OBD indication, however, is seen as having the larger market opportunity, and Progenics may have the edge in its development program, analysts said.
Jonathan Aschoff, an analyst with Brean, Murray & Carret, wrote in a research note that, "Although GSK still retains rights to Entereg for postoperative ileus, we believe chronic OBD is by far the largest opportunity for an opioid receptor antagonist. Therefore, any delay in initiating the next Entereg trial in OBD will allow Progenics to take a lead and weaken the competitive advantage of Entereg." Adolor's Entereg currently carries a "black box" warning for safety reasons, and could pose an issue for Entereg in the larger OBD market. "If Progenics takes the lead, we believe the cleaner toxicity profile of Relistor will make it even more challenging for Entereg to compete in the chronic OBD market," Aschoff said.
Progenics' injectable Relistor currently is approved for constipation resulting from the use of powerful painkillers by terminally ill patients. Tarrytown, N.J.-based Progenics and partner Wyeth are studying an oral formulation of Relistor for a similar indication, known as opioid-induced constipation.
A Phase III trial of Relistor in patients with postoperative ileus did not meet any of the study goals. But a Phase II study of oral Relistor showed some benefit in treating opioid-induced constipation in patients with chronic, non-malignant pain. (See BioWorld Today, May 23. 2008.)
Carr, the Needham analyst, said it was "not a vote of confidence" that GSK walked away from Adolor's OBD program. A spokesman for GSK did not immediately return a call seeking comment on the decision.
GSK also has returned to Adolor the rights related to Entereg for irritable bowel syndrome and nonopioid-induced forms of constipation or bowel dysfunction. There have been no active development programs for those indications, Adolor said.
GSK indicated earlier it was evaluating Entereg, and the market did not seem surprised by the decision on the OBD indication. Shares in Adolor (NASDAQ:ADLR) hardly moved, with an increase of 1 cent, closing at $3.27. Shares in Progenics (NASDAQ: PGNX) were up 53 cents, closing at $14.27.
mfg ipollit
http://www.tmcnet.com/usubmit/2008/09/02/3629494.htm
September 02, 2008
GSK Hands Entereg OBD Indication Back to Adolor
(BioWorld Today Via Acquire Media NewsEdge) Drug giant GlaxoSmithKline Plc handed back rights to bowel drug Entereg to Adolor Corp., a setback for GSK's partner but good news for Progenics Pharmaceuticals Inc., which has its own bowel drug Relistor that could compete with Entereg. Both Adolor and Progenics are seeking to expand use of their respective FDA-approved bowel drugs.
But Adolor's plans could be delayed as a result of GSK walking away from a program evaluating Entereg in chronic opioid-induced bowel dysfunction. Meantime, Progenics could move out front, with a Phase III study of its bowel drug expected next year.
"Anything that slows down Entereg will be good for Relistor," Need ham & Co. analyst Alan Carr told BioWorld Today. Carr, who tracks Progenics, said the setback for Adolor's Entereg gives Progenics "more breathing room."
Adolor will regain the rights to Entereg in opioid bowel dysfunction, or OBD, and will likely need to find another partner to advance Entereg in that indication. Now that the FDA has lifted a clinical hold on Entereg for OBD, Adolor is considering conducting another study under a special protocol assessment.
Michael R. Dougherty, president and CEO of Adolor, said in a statement that "Adolor maintains a portfolio of development candidates that may potentially serve this patient population, including Entereg, our combination product program, and additional earlier stage compounds. We intend now to explore discussions with potential partners regarding this portfolio, and to submit to the FDA for review a protocol for an additional study of Entereg in OBD under a special protocol assessment."
Adolor's Entereg currently is approved for postoperative ileus, in which the bowels lose their motility. GSK is retaining rights to Entereg for postoperative ileus (POI), and the companies will continue to collaborate on the development and commercialization of Entereg for POI in the US., Adolor said.
The OBD indication, however, is seen as having the larger market opportunity, and Progenics may have the edge in its development program, analysts said.
Jonathan Aschoff, an analyst with Brean, Murray & Carret, wrote in a research note that, "Although GSK still retains rights to Entereg for postoperative ileus, we believe chronic OBD is by far the largest opportunity for an opioid receptor antagonist. Therefore, any delay in initiating the next Entereg trial in OBD will allow Progenics to take a lead and weaken the competitive advantage of Entereg." Adolor's Entereg currently carries a "black box" warning for safety reasons, and could pose an issue for Entereg in the larger OBD market. "If Progenics takes the lead, we believe the cleaner toxicity profile of Relistor will make it even more challenging for Entereg to compete in the chronic OBD market," Aschoff said.
Progenics' injectable Relistor currently is approved for constipation resulting from the use of powerful painkillers by terminally ill patients. Tarrytown, N.J.-based Progenics and partner Wyeth are studying an oral formulation of Relistor for a similar indication, known as opioid-induced constipation.
A Phase III trial of Relistor in patients with postoperative ileus did not meet any of the study goals. But a Phase II study of oral Relistor showed some benefit in treating opioid-induced constipation in patients with chronic, non-malignant pain. (See BioWorld Today, May 23. 2008.)
Carr, the Needham analyst, said it was "not a vote of confidence" that GSK walked away from Adolor's OBD program. A spokesman for GSK did not immediately return a call seeking comment on the decision.
GSK also has returned to Adolor the rights related to Entereg for irritable bowel syndrome and nonopioid-induced forms of constipation or bowel dysfunction. There have been no active development programs for those indications, Adolor said.
GSK indicated earlier it was evaluating Entereg, and the market did not seem surprised by the decision on the OBD indication. Shares in Adolor (NASDAQ:ADLR) hardly moved, with an increase of 1 cent, closing at $3.27. Shares in Progenics (NASDAQ: PGNX) were up 53 cents, closing at $14.27.
mfg ipollit
Progenics hat heute auch eine PI eines AKs gegen Prostatakebs gestartet, der auf der AK-Technologie von Seattle Genetics basiert...
September 08, 2008 07:00 AM Eastern Daylight Time
Progenics Initiates Phase 1 Clinical Study of Targeted Therapy for Prostate Cancer
Monoclonal Antibody-Drug Conjugate Selectively Targets Cancer Cells
TARRYTOWN, N.Y.--(BUSINESS WIRE)--Progenics Pharmaceuticals, Inc. (Nasdaq: PGNX) today announced the initiation of a phase 1 dose-escalation clinical study of its prostate-specific membrane antigen (PSMA) antibody-drug conjugate (ADC). PSMA ADC is an investigational therapy that combines a prostate-cancer antibody with a cancer drug. Unlike traditional chemotherapy, PSMA ADC is designed to deliver the drug selectively to prostate cancer cells by targeting a surface protein, PSMA. The clinical study will include patients with progressive, metastatic, hormone-refractory disease following prior taxane chemotherapy, and will assess the safety, tolerability and initial clinical activity of PSMA ADC followed by the option to continue therapy for a total of 12 months.
“In pre-clinical studies, PSMA ADC has exhibited a high level of tumor-specific activity,” said Paul J. Maddon, M.D., Ph.D., Progenics Pharmaceuticals’ Founder, Chief Executive Officer and Chief Science Officer. “Now, our first-in-man clinical studies will assess initial anti-tumor activity in patients after 12 weeks and subsequently, with an optional follow-up period, provide up to one year of dosing information.”
Antibody-drug conjugate therapy
PSMA ADC is an antibody-drug conjugate that consists of a fully human monoclonal antibody that binds PSMA and is linked to a highly potent cancer drug, a derivative of auristatin.
The monoclonal antibody portion of PSMA ADC selectively targets PSMA, a protein that is abundantly expressed on the surface of prostate cancer cells. Using technology licensed from Seattle Genetics, Inc. (Nasdaq: SGEN), the fully human PSMA antibody is linked to a potent derivative of auristatin, a cancer drug that inhibits cell proliferation by disrupting the cellular “backbone” (i.e., microtubules) required for replication. The resultant antibody-drug conjugate attaches to the PSMA protein on the surface of prostate cancer cells and is designed to:
internalize the antibody-drug conjugate into the cancer cell;
release and thereby activate the cancer drug; and
destroy the malignant cell.
The cancer drug is chemically linked to the antibody and is designed to activate only after the antibody recognizes a cancer cell.
Two-stage clinical study design
The phase 1, open-label, dose-escalation clinical trial will include as many as 40 men with progressive, hormone-refractory prostate cancer, and who had prior therapy with taxane chemotherapy drugs. The study will investigate the duration of clinical benefit derived from PSMA ADC treatment while also assessing the drug’s safety and tolerability. The initial 12-week period will evaluate up to four intravenous doses of PSMA ADC over five dose levels, administered at three-week intervals. The study will include evaluations of pharmacodynamics, radiographic changes in tumor burden, and changes in prostate-specific antigen (PSA) and circulating tumor cell (CTC) values compared to baseline.
Following the 12-week period, patients will be offered, at their physician’s discretion, the option to continue treatment for an additional 39 weeks with the same dose of PSMA ADC as administered in their initial cohort. Qualified subjects will receive up to 13 additional doses of study drug at three-week intervals.
Pre-clinical results
A pre-clinical study, presented at the AACR Annual Meeting in April, demonstrated that treatment with PSMA ADC improved survival in a mouse model of androgen-independent prostate cancer. Post-tumor implantation, the median survival for control mice was 57 days. In contrast, nine of 10 animals in the PSMA ADC treatment group survived until study end at 150 days. Notably, of these nine mice, seven had no measurable tumor at day 150. No overt toxicity was observed with PSMA ADC treatment in this preclinical animal model.
About PSMA
PSMA is a protein abundantly expressed on the surface of prostate cancer cells and on the neovasculature of many types of solid tumors. PSMA expression is increased in high-grade, metastatic and hormone-refractory prostate cancer. Since PSMA has limited expression on normal tissues, it is an excellent therapeutic target.
Progenics is engaged in two programs that target PSMA for the treatment of prostate cancer. In addition to developing PSMA ADC, the Company's second program pursues vaccines that are designed to augment the body's ability to recognize and eradicate cancer cells that express PSMA.
mfg ipollit
September 08, 2008 07:00 AM Eastern Daylight Time
Progenics Initiates Phase 1 Clinical Study of Targeted Therapy for Prostate Cancer
Monoclonal Antibody-Drug Conjugate Selectively Targets Cancer Cells
TARRYTOWN, N.Y.--(BUSINESS WIRE)--Progenics Pharmaceuticals, Inc. (Nasdaq: PGNX) today announced the initiation of a phase 1 dose-escalation clinical study of its prostate-specific membrane antigen (PSMA) antibody-drug conjugate (ADC). PSMA ADC is an investigational therapy that combines a prostate-cancer antibody with a cancer drug. Unlike traditional chemotherapy, PSMA ADC is designed to deliver the drug selectively to prostate cancer cells by targeting a surface protein, PSMA. The clinical study will include patients with progressive, metastatic, hormone-refractory disease following prior taxane chemotherapy, and will assess the safety, tolerability and initial clinical activity of PSMA ADC followed by the option to continue therapy for a total of 12 months.
“In pre-clinical studies, PSMA ADC has exhibited a high level of tumor-specific activity,” said Paul J. Maddon, M.D., Ph.D., Progenics Pharmaceuticals’ Founder, Chief Executive Officer and Chief Science Officer. “Now, our first-in-man clinical studies will assess initial anti-tumor activity in patients after 12 weeks and subsequently, with an optional follow-up period, provide up to one year of dosing information.”
Antibody-drug conjugate therapy
PSMA ADC is an antibody-drug conjugate that consists of a fully human monoclonal antibody that binds PSMA and is linked to a highly potent cancer drug, a derivative of auristatin.
The monoclonal antibody portion of PSMA ADC selectively targets PSMA, a protein that is abundantly expressed on the surface of prostate cancer cells. Using technology licensed from Seattle Genetics, Inc. (Nasdaq: SGEN), the fully human PSMA antibody is linked to a potent derivative of auristatin, a cancer drug that inhibits cell proliferation by disrupting the cellular “backbone” (i.e., microtubules) required for replication. The resultant antibody-drug conjugate attaches to the PSMA protein on the surface of prostate cancer cells and is designed to:
internalize the antibody-drug conjugate into the cancer cell;
release and thereby activate the cancer drug; and
destroy the malignant cell.
The cancer drug is chemically linked to the antibody and is designed to activate only after the antibody recognizes a cancer cell.
Two-stage clinical study design
The phase 1, open-label, dose-escalation clinical trial will include as many as 40 men with progressive, hormone-refractory prostate cancer, and who had prior therapy with taxane chemotherapy drugs. The study will investigate the duration of clinical benefit derived from PSMA ADC treatment while also assessing the drug’s safety and tolerability. The initial 12-week period will evaluate up to four intravenous doses of PSMA ADC over five dose levels, administered at three-week intervals. The study will include evaluations of pharmacodynamics, radiographic changes in tumor burden, and changes in prostate-specific antigen (PSA) and circulating tumor cell (CTC) values compared to baseline.
Following the 12-week period, patients will be offered, at their physician’s discretion, the option to continue treatment for an additional 39 weeks with the same dose of PSMA ADC as administered in their initial cohort. Qualified subjects will receive up to 13 additional doses of study drug at three-week intervals.
Pre-clinical results
A pre-clinical study, presented at the AACR Annual Meeting in April, demonstrated that treatment with PSMA ADC improved survival in a mouse model of androgen-independent prostate cancer. Post-tumor implantation, the median survival for control mice was 57 days. In contrast, nine of 10 animals in the PSMA ADC treatment group survived until study end at 150 days. Notably, of these nine mice, seven had no measurable tumor at day 150. No overt toxicity was observed with PSMA ADC treatment in this preclinical animal model.
About PSMA
PSMA is a protein abundantly expressed on the surface of prostate cancer cells and on the neovasculature of many types of solid tumors. PSMA expression is increased in high-grade, metastatic and hormone-refractory prostate cancer. Since PSMA has limited expression on normal tissues, it is an excellent therapeutic target.
Progenics is engaged in two programs that target PSMA for the treatment of prostate cancer. In addition to developing PSMA ADC, the Company's second program pursues vaccines that are designed to augment the body's ability to recognize and eradicate cancer cells that express PSMA.
mfg ipollit
zu Exelixis...
http://seekingalpha.com/article/93716-exelixis-a-platform-co…
Exelixis: A Platform Company for Oncology
by: Ohad Hammer posted on: September 03, 2008 | about stocks: EXEL
The pharmaceutical industry is in the midst of a severe innovation crisis, where more R&D money results in less approved drugs. In parallel, sales of most traditional blockbusters are being cannibalized by generic competition, forcing the big pharmas to exploit new concepts and rush new drug candidates to their pipelines.
Nearly half of all drugs that are expected to enter the clinic in the coming years will be aimed at the oncology market. Consequently, early stage drug development for cancer is going to be the most active area in the industry, with an abundance of investment opportunities, particularly in small and medium companies.
Many investors prefer to stay away from oncology drug development, where success rates are at a worrying low, hovering around 5% compared to 10% for the whole industry. Nevertheless, among the hundreds of publicly traded companies which are engaged in oncology, there may be a group of companies which have better chances of succeeding in such a monumental task. These companies are what I like to call platform companies.
In this article, I will try to explain what makes a good platform company and why Exelixis (EXEL) can be regarded as a good (yet not a perfect) example. For the sake of clarification, I certainly do not claim that platform companies are the only investment-worthy biotech companies, however, when it comes to early stage drug development, platform companies are an excellent place to start.
In general, platform companies can be evaluated based on five criteria:
1) A validated platform
Having a validated drug discovery platform is the first and most important criterion for defining a good platform company. The platform is typically comprised of a combination of technology, experienced personnel and intellectual property that can generate a stream of drug candidates. Most importantly, investing should be done only after a product of the platform demonstrates activity in clinical trials. Having a clinically validated product is not a guarantee for future success of the platform nor does it mean that the specific agent will reach the market, but it does imply that one or more of the platform’s products stand a reasonable chance of becoming a commercial drug. A validated platform may increase overall success rates, yet the odds of a particular drug candidate to make it all the way to approval are still low.
It is hard to define exactly what “clinical validation” means, but there should be a clear response in the form of disease control, tumor shrinkage or side effects, preferably with more than one candidate, in patients with limited treatment options. A good example for a platform validation may be Immunogen’s (IMGN) antibody-drug conjugation platform, which empowers antibodies by conjugating them to toxic drug payloads. T-DM1 is comprised of the blockbuster antibody, Herceptin, empowered by Immunogen’s technology. It had remarkable activity in two clinical trials in patients who had progressed while receiving Herceptin, and therefore can be regarded as a good clinical validation of Immunogen’s technology.
2) Specialization and Differentiation
The field of drug development is becoming more rich and versatile on the one hand, but also more complex and competitive on the other. Instead of relying on traditional trial and error and serendipity for discovering drugs, the industry is now moving to rational design and computerized tools, which require a great deal of expertise. This is why small companies must not spread their resources too thinly but instead focus on one field. By specializing in a certain area, and developing a deep understanding of the underlying science that stands at the basis of development, even a small company can achieve a competitive edge over the rest of the market.
The ability to block new entrants to the field by means of intellectual property is always an advantage, as clearly demonstrated in the case of Alnylam (ALNY) and Isis Pharmaceuticals (ISIS), which became the gate-keepers of their fields with the help of a draconian patent portfolio. Timing is also a crucial issue in finding a niche to specialize in, and since the process of developing a drug is a linear and lengthy one, a several years head start is needed in most cases.
3) Multiple shots on goal
A good platform company always has multiple agents in active development simultaneously, preferably from the same validated platform. No matter how promising a particular compound looks, it is imperative to remember that most drugs fail, including those that demonstrate spectacular results in early clinical trials; thus, banking on one or two early stage compounds is statistically futile.
Just to make it clear, active development does not include just having the compounds listed on the pipeline chart, waiting to fill the gap created by discontinuation of programs, but a constant pursuit of the compound’s development with the ultimate goal of advancing it into multiple clinical trials. Due to the enormous costs associated with drug development, small and medium companies cannot finance more than a handful of programs, which leads us to the next point.
4) Deep Pocketed Partners
Partnership deals with large pharmaceutical companies are the only viable options for a small company to obtain a broad pipeline. Collaborating with large partners serves two prime purposes. First, it allows small players to survive the time needed to develop drugs without infinite dilution. In addition, any partnership deal is perceived as a vote of confidence for the technology from a savvy player in the industry, which bodes well for investors’ confidence and valuation.
The dark side of partnership deals is that if the drug hits the market, the large partner enjoys most of the gains, but in most cases the risks and dilution accompanied with developing the drug independently are far graver. Trying to keep all the promising candidates in-house can turn these assets into a huge liability.
Small biotechs must realize what they are good at, feeding the industry with a large number of candidates, while letting the big pharmas do what they are best at, commercializing products. The most elegant illustration of this approach is Isis, which declares itself as a “fully disintegrated” company that does not intend to be involved in late stage trials and marketing. Although Isis still hasn’t enjoyed meaningful sales of its products, the number of programs it has and its pipeline diversification are overwhelming.
Isis, a $1.5B company with thirteen drugs in the clinic and an expected operating loss of only $25 million in 2008, is also one of the few development stage companies that may reach profitability without actual royalties on sales.
5) Field Of Activity
Evaluating the field in which a company operates is somewhat trickier than the other criteria, as it is hard to define the different parameters and the balance between them. These parameters include validation and prior success rates of the mechanism of action and the target, the competitive landscape, entry barriers and intellectual property. Because a validated field usually comes with plenty of competition (from drugs in the market and in the clinic), it is very hard to find a platform company that is optimally positioned.
Companies with a good blend can typically be divided into pioneers like Isis and Alnylam, whose technologies are still not commercialized and “next-generation” companies such as Immunogen and Seattle Genetics (SGEN) that improve a validated class of drugs. Isis and Alnylam are high risk companies because of the lack of complete validation of their fields, but they have a considerable gap over the rest of the market. Immunogen’s and Seattle Genetics’ markets are more mature and proven, but the companies are facing more competition.
It is plain to see that the five criteria are intertwined in a way that forces companies to combine them. It is impossible to have multiple candidates in the clinic without multiple collaboration deals, which are hard to strike without a good platform that can produce multiple promising candidates. Partnership deals often lead to an increase in the attention a specific area receives, but such deals may very well be a result of a clinical validation by a single drug candidate, which in turn, may lead to the validation of an entire field.
Exelixis as a Platform Company
Exelixis is active in the ever growing market of kinase inhibitors (KIs) for the treatment of cancer, that is, drugs that block the activity of kinases in cancer cells. Cancer cells are often described as cells that are out of control: They proliferate quickly, ignore death signals, invade nearby tissues and eventually metastasize to distant organs. Disease onset and advancement are associated with processes such as cell growth, motility and blood-vessel formation, which are governed by a complex network made of kinases. Thus, blocking these processes by inhibiting the relevant kinases has emerged as one of the most attractive approaches to fighting cancer.
Together with monoclonal antibodies, kinase inhibitors represent a paradigm shift in cancer treatment from cytotoxic agents to targeted therapies, a trend that is constantly growing. Like antibodies for cancer, kinase inhibitors target tumors while sparing healthy cells and consequently lead to better activity with fewer side effects.
Kinase inhibitors, however, possess several advantages over antibodies. The most evident advantage is that KIs can hit targets inside the cell while antibodies can only bind targets presented on the cell surface, so internal targets are approachable only by KIs. Another advantage is the fact that KIs can be given orally, which is a major factor in terms of patient convenience, especially given the typical long treatment duration associated with targeted therapies. Another advantage, which will be later discussed in the article, is the ability to produce KIs that hit several targets at once.
The commercial potential of kinase inhibitors for cancer was validated by the approval of Novartis’ (NVS) Gleevec in 2001. Gleevec, which still remains the best selling kinase inhibitor drug, is considered one of the biggest success stories of modern medicine, owing to its long lasting benefit and excellent safety profile. Naturally, Gleevec’s success spurred considerable efforts to develop additional kinase inhibitors for cancer.
Seven years later, the field of kinase inhibitors is as active as ever, with eight additional approved drugs (three of which were launched only last year) and over a hundred kinase inhibitors in the clinic, most of which are evaluated for oncology indications. The implication of kinases in cancer is constantly being elucidated and today it is clear that they play a cardinal role in virtually every type of cancer. Commercial-wise, the market of kinase inhibitors is poised to grow at a fast pace thanks to new approvals as well as label extensions for marketed drugs.

It should be noted that although the benefit of kinase inhibitors is undeniable, Gleevec’s success in terms of duration of response and safety profile has not been replicated by other agents. In fact, many patients eventually develop resistance even against Gleevec.
The arms race between cancer and kinase inhibitors is just beginning, and the industry is placing its hopes on new approaches to developing better drugs. Some of these approaches include identifying new targets as well as new binding sites within known targets, simultaneously inhibiting a target with more than one drug, tailoring drugs for well defined populations and inhibiting several targets at once.
Examining Exelixis based on four of the five criteria - field of activity, a validated platform, multiple candidates and large partners makes it look very promising. The KI field is a highly validated field with plenty of growth ahead, so investing in this field seems like a smart thing to do.
With respect to clinical validation of the platform, several of Exelixis’ compounds have shown activity in humans. Although this activity has been shown in preliminary trials, it seems that Exelixis has one of the most promising pipelines in the industry. There are obviously no problems on the partnerships front, as Exelixis is involved with some high profile names from the likes of GlaxoSmithKline (GSK), Genentech (DNA), Bristol-Myers Squibb (BMY) and Wyeth (WYE). In terms of number of compounds in clinical trials, Exelixis has 11 drug candidates in over 20 clinical trials, which is quite impressive for a company its size.
The biggest issue with any KI company is the challenging competitive landscape. Because the field is so promising, it also became extremely crowded and competitive. There is no “top dog” in kinase inhibitors, but rather there are dozens of participants, from small biotechs to the world’s largest pharmaceutical companies, which naturally raises the question whether a single company can have a competitive edge over the rest of the market.
A company that develops kinase inhibitors typically does not have the luxury of blocking competitors by means of intellectual property, as all players are relatively free to operate in terms of the targets they explore, the way the compound binds the target, the approach for screening compounds and so on. Nevertheless, it would also be too simplistic to view this industry as a herd of “me too” companies, as there is still room for each company to differentiate itself.
Exelixis managed to differentiate itself based on three factors.
1) The Biggest Small Company
Exelixis is a small pure-play company focused entirely on kinase inhibitors. The risks of investing in small cap biotechs are evident, but so are the benefits. Exelixis provides 100% exposure to the field of kinase inhibitors, with a strong emphasis on cancer. This is something investors will not find in any of the larger players, no matter how active they are, not to mention the innovation and agility that are often absent in large pharmas.
For some reason, there is an inverse relationship between the innovation and the productivity of a company and its size. Smaller companies tend to be more productive in early stage drug development due to creative thinking and better maximization of resources. When it comes to the costly clinical stage, though, the large companies have the luxury of pursuing several paths in parallel as well as the capability of coping with the logistics and regulatory challenges.
One advantage Exelixis has over the myriad of small biotechs is the diversity and scope of its pipeline. As can be seen in the table below, in terms of the number of compounds in clinical development, Exelixis is in the big leagues.
Notably, a comparison based on number of drugs in the pipeline is flawed because it does not say anything about the quality of these drugs, nor does it specify the different trials with every compound. For example, Exelixis has one candidate (XL184) in phase III for a very small indication as well as in three earlier trials, while Pfizer’s phase III compounds (Sutent and axitinib) are being evaluated in tens of trials, including several phase III trials for very large indications.
In addition, being a small company, several of Exelixis’ compounds are partnered, as opposed to the large players who own the majority of their portfolios. Nevertheless, as one-dimensional as this comparison may be, it illustrates the fact that Exelixis’ pipeline is focused in terms of the therapeutic area, yet very diverse in terms of the number of compounds and the targets they hit. Besides, the impact of 15% of product sales (in the form of royalties) on a company like Exelixis is substantially stronger than that of 85% of sales on a large pharmaceutical company.

2) Discrete Patient Populations
It has been well recognized that classifying patients based on the origin of the tumor (i.e. pancreatic, breast cancer) ignores countless other important differences in the biology of the disease. Today, most cancer drugs are primarily given based on the site of tumor origin and consequently lead to a wide variability in response, both in terms of benefits and side effects. In order to optimally treat cancer, patients should be classified based on additional factors, with a strong emphasis on the mechanisms involved in their disease.
Distinguishing between the different subtypes is instrumental in the case of targeted therapies such as kinase inhibitors and monoclonal antibodies, because these agents usually affect one or a few targets that may not be relevant for all patients. Genentech’s Herceptin, for instance, has no effect on 70-80% of breast cancer patients, but leads to a strong benefit in the rest of the patients, whose tumors utilize Herceptin’s target (Her2) in order to grow and advance. Had Herceptin (over $5 billion in sales estimated this year) been evaluated in unselected breast cancer patients, it would probably have never reached the market due to lack of efficacy.
The commercial potential of niche markets is, by definition, more limited, but the strategy of addressing discrete patient populations holds several key advantages over starting with larger populations. First and foremost, focusing on small, well defined populations can lead to overwhelming results in terms of activity, and consequently increase the likelihood of approval. Moreover, it may provide early validation of the target or the compound’s activity in humans, which decreases the risk and renders the compound more attractive to potential partners.
Second, the development process involves smaller trials, so it is faster and cheaper than that of large indications. Third, niche markets are less crowded with competitors, and often ignored by the large pharmas. Fourth, although the addressable market is more modest, the drug can generate recurring revenues that are desperately needed by small companies. Fifth, assuming that the target population is well defined and highly responsive to the drug, revenues per patient are higher compared to the average in the industry, both due to longer treatment periods and patients’ and insurers’ willingness to pay more if they know there is a high likelihood of long-lasting benefit.
This has led many companies to realize that it might be better to focus on a subset of patients instead of targeting the whole market. Exelixis is spearheading the pursuit of personalized medicine by examining the pathways tumors “hijack” in order to grow, and identifying the right patient populations that are likely to benefit from a particular compound’s spectrum of activity.
The company’s compounds hit targets that may be relevant across a wide range of malignancies, representing very large target markets. Nevertheless, the company made a strategic decision to initially pursue niche markets with limited treatment options and use approvals in these small markets as a stepping stone for additional indications. Such niche markets might be either rare cancers or well defined subsets of patients within a large population of patients. This strategy is already starting to bear fruit, judging by results from two candidates: XL184 and XL880.
XL184 entered the clinic in 2005 and is currently being evaluated in three indications. Its shortest route to market is an ongoing phase III trial in 315 medullary thyroid cancer [MTC] patients, as it could hit the market as early as 2011. XL184 was active in all advanced MTC patients in a recently published trial, so chances for approval are high. Advanced MTC is considered a highly unmet medical need but the market size for this indication is very small (less than 1000 patients in the US and Europe every year). Thus, the overall potential for this indication amounts to several tens of millions annually, but it can serve as a proof of concept for the drug’s activity and safety profile, decreasing the overall risk in additional programs.
XL880 is currently being developed in collaboration with GSK. In a recently published phase II trial, XL880 showed impressive activity in patients with papillary renal cancer. These patients, which account for 8-10% of kidney cancer cases, represent an attractive niche because they seem to be less responsive to the approved targeted agents for kidney cancer. In the phase II trial, XL880 demonstrated impressive activity in papillary renal cancer patients with an exceptional duration of responses, which may lead to a registration trial already next year, only four years after entering the clinic.
The immediate target population for this indication is estimated at 10,000 patients in the United States and Europe. Importantly, because XL880’s benefit in the phase II trial lasted, on average, more than a year, the commercial opportunity can be as high as $400M annually.
3) Multi-Targeted Approach
The concept of hitting a combination of kinases simultaneously has been gaining momentum in the industry, given the complexity and multi-factorial nature of the cancer. In order to develop and advance, cancer cells must acquire multiple distinct capabilities such as the capability to proliferate, resist death signals, create new blood vessels and invade nearby tissues. Each capability can be governed by multiple pathways which work independently, in parallel, and through interconnections to promote cancer development. Therefore, by hitting multiple targets at once, a compound may employ more than one mechanism of action and increase spectrum of activity.
Multi-targeted kinase inhibitors can also target several points in the same pathway for achieving a more pronounced effect and delay the emergence of drug resistance. There may also be a commercial value in multi-targeted KIs because one drug can theoretically replace two or three single targeted agents, thereby lowering the cost of treatment.
In recent years, three drugs thought to inhibit multiple kinases entered the market (Sutent, Nexavar and Tykerb), and are seen as a validation of this approach. Interestingly, even kinase inhibitors which were developed as single kinase inhibitors, often have some degree of activity against additional kinases, whether clinically relevant or not. A good example for that might be Gleevec, which had started as a single kinase inhibitor, but then was reborn as an inhibitor of at least two other kinases. At the moment, Exelixis seems to favor the multi-kinase inhibitors approach, with 7 multi-targeted and 4 single-targeted kinase inhibitors in the clinic.
Although there is a wide consensus that simultaneous inhibition of multiple targets may optimize the use of targeted therapies, some question the benefit of multi-targeted inhibitors pointing at several issues. Manipulating several targets at once adds more complexity, on top of the intrinsic complexity of developing a drug against one target. More targets means more variables and uncertainty in terms of the choosing the right combination, the degree of inhibition of each target and the safety profile.
The promiscuous activity of multi-targeted kinase inhibitors often leads to increased toxicity, so these agents are not likely to have an ideal safety profile and consequently may not be given in sufficiently high doses. In addition, it is not always clear whether the clinical activity is due to inhibition of one or multiple targets, and the role each target plays. Most of Nexavar’s activity, for instance, is probably not the result of inhibition of the initial primary target for which the compound was developed, but due to a secondary target. As a result, there are some who believe that simultaneous inhibition of multiple targets should be done with several single-targeted agents rather than with one multi-targeted agent.
Like everything else in drug development, there is no clear solution for this debate, as there is probably more than one way of fighting cancer with kinase inhibitors. The right approach should be chosen on a case by case basis, depending on the specific drug, patient characteristics and the target itself.
Notwithstanding this debate, choosing the right targets is the first, and perhaps the most important step in the process. There is a tradeoff between the extent of clinical validation and the competitive landscape for each target.
Developing drugs that hit validated targets decreases the risk of failure on the one hand, but also involves fierce competition from approved and investigational agents. Examples for two highly validated targets may be EGFR (targeted by Tarceva and Iressa) and Bcr-Abl (targeted by Gleevec, Sprycel and Tasigna). Dethroning an approved drug is a challenging task, not to mention potential competition from compounds in development, so even if a company has a drug that works, it will not necessarily succeed in getting market share.
It is important to understand, though, that presence of approved agents does not necessarily hamper the potential of new compounds. Acknowledging that advanced cancer is almost impossible to cure, the industry is now talking about making it a chronic disease by using many lines of therapy. Kinase inhibitors’ inability to cure the disease and the emergence of resistance creates the need for developing second and third lines of therapy as well as combination treatments. There is, for instance, room for EGFR inhibitors that are active in Tarceva resistant patients or can be given in combination with Tarceva to improve activity and delay the onset of resistance.
Targets for which there are no marketed drugs involve more risk, but also represent the potential of opening up new markets with little or no competition. Exelixis decided to focus on kinases that are still not clinically validated, but such that have a clear role in cancer biology, backed up by a large body of scientific evidence. These targets include MET, PI3K, Hedgehog and MEK. Obviously, there are plenty of other compounds in development that target these kinases, and it is still premature to assess how Exelixis will fare with the competition, but thanks to its size and focus, it managed to be at the forefront of these exciting field.
Exelixis’s approach of targeting the right patient populations and the fact that some of its compounds inhibit multiple kinases may help it differentiate its compounds. In some cases, Exelixis further decreases the risk by developing two distinct compounds against the same target. For example, XL147 and XL765 both inhibit PI3K but the latter inhibits an additional kinase in the same signaling pathway - mTOR. On top of being an independent attempt of striking PI3K, each compound represents a different approach for inhibiting the same pathway in terms of the ideal number of targets to hit in this pathway. It is still unclear which approach is superior, and in which cases, so pursuing both routes in parallel sounds like a good idea.
Summary
Exelixis operates in one of the most promising yet competitive markets in the pharmaceutical industry, where there are always multiple candidates against any given target. The company is not nor will it ever be the undisputed leader of the KI market; however, it managed to distinguish itself as a hybrid between a small biotech and a large pharmaceutical company.
On the one hand, it is an extremely focused company with one of the leading R&D teams in the industry. On the other, it has a broad and diverse pipeline of promising compounds that may be applicable to a wide array of cancers. Most of these compounds will fail either due to poor performance or competing agents, but overall, Exelixis should have enough shots on goal in order to make it.
Disclosure: Author is long EXEL, ISIS, IMGN & SGEN.
***********
The cash issue is the most important factor in thenear term, no doubt. I will try to touch on that matter in the future, but that's an issue everybody including company's mangement is aware of. I expect they'll start to monetizing their portfolio in Q4, after the GSK decision(s). I hear they are very close to siging a deal for one of their PI3K compounds, which are not part the GSK collaboration.
mfg ipollit" target="_blank" rel="nofollow ugc noopener">http://seekingalpha.com/article/93716-exelixis-a-platform-co…
Exelixis: A Platform Company for Oncology
by: Ohad Hammer posted on: September 03, 2008 | about stocks: EXEL
The pharmaceutical industry is in the midst of a severe innovation crisis, where more R&D money results in less approved drugs. In parallel, sales of most traditional blockbusters are being cannibalized by generic competition, forcing the big pharmas to exploit new concepts and rush new drug candidates to their pipelines.
Nearly half of all drugs that are expected to enter the clinic in the coming years will be aimed at the oncology market. Consequently, early stage drug development for cancer is going to be the most active area in the industry, with an abundance of investment opportunities, particularly in small and medium companies.
Many investors prefer to stay away from oncology drug development, where success rates are at a worrying low, hovering around 5% compared to 10% for the whole industry. Nevertheless, among the hundreds of publicly traded companies which are engaged in oncology, there may be a group of companies which have better chances of succeeding in such a monumental task. These companies are what I like to call platform companies.
In this article, I will try to explain what makes a good platform company and why Exelixis (EXEL) can be regarded as a good (yet not a perfect) example. For the sake of clarification, I certainly do not claim that platform companies are the only investment-worthy biotech companies, however, when it comes to early stage drug development, platform companies are an excellent place to start.
In general, platform companies can be evaluated based on five criteria:
1) A validated platform
Having a validated drug discovery platform is the first and most important criterion for defining a good platform company. The platform is typically comprised of a combination of technology, experienced personnel and intellectual property that can generate a stream of drug candidates. Most importantly, investing should be done only after a product of the platform demonstrates activity in clinical trials. Having a clinically validated product is not a guarantee for future success of the platform nor does it mean that the specific agent will reach the market, but it does imply that one or more of the platform’s products stand a reasonable chance of becoming a commercial drug. A validated platform may increase overall success rates, yet the odds of a particular drug candidate to make it all the way to approval are still low.
It is hard to define exactly what “clinical validation” means, but there should be a clear response in the form of disease control, tumor shrinkage or side effects, preferably with more than one candidate, in patients with limited treatment options. A good example for a platform validation may be Immunogen’s (IMGN) antibody-drug conjugation platform, which empowers antibodies by conjugating them to toxic drug payloads. T-DM1 is comprised of the blockbuster antibody, Herceptin, empowered by Immunogen’s technology. It had remarkable activity in two clinical trials in patients who had progressed while receiving Herceptin, and therefore can be regarded as a good clinical validation of Immunogen’s technology.
2) Specialization and Differentiation
The field of drug development is becoming more rich and versatile on the one hand, but also more complex and competitive on the other. Instead of relying on traditional trial and error and serendipity for discovering drugs, the industry is now moving to rational design and computerized tools, which require a great deal of expertise. This is why small companies must not spread their resources too thinly but instead focus on one field. By specializing in a certain area, and developing a deep understanding of the underlying science that stands at the basis of development, even a small company can achieve a competitive edge over the rest of the market.
The ability to block new entrants to the field by means of intellectual property is always an advantage, as clearly demonstrated in the case of Alnylam (ALNY) and Isis Pharmaceuticals (ISIS), which became the gate-keepers of their fields with the help of a draconian patent portfolio. Timing is also a crucial issue in finding a niche to specialize in, and since the process of developing a drug is a linear and lengthy one, a several years head start is needed in most cases.
3) Multiple shots on goal
A good platform company always has multiple agents in active development simultaneously, preferably from the same validated platform. No matter how promising a particular compound looks, it is imperative to remember that most drugs fail, including those that demonstrate spectacular results in early clinical trials; thus, banking on one or two early stage compounds is statistically futile.
Just to make it clear, active development does not include just having the compounds listed on the pipeline chart, waiting to fill the gap created by discontinuation of programs, but a constant pursuit of the compound’s development with the ultimate goal of advancing it into multiple clinical trials. Due to the enormous costs associated with drug development, small and medium companies cannot finance more than a handful of programs, which leads us to the next point.
4) Deep Pocketed Partners
Partnership deals with large pharmaceutical companies are the only viable options for a small company to obtain a broad pipeline. Collaborating with large partners serves two prime purposes. First, it allows small players to survive the time needed to develop drugs without infinite dilution. In addition, any partnership deal is perceived as a vote of confidence for the technology from a savvy player in the industry, which bodes well for investors’ confidence and valuation.
The dark side of partnership deals is that if the drug hits the market, the large partner enjoys most of the gains, but in most cases the risks and dilution accompanied with developing the drug independently are far graver. Trying to keep all the promising candidates in-house can turn these assets into a huge liability.
Small biotechs must realize what they are good at, feeding the industry with a large number of candidates, while letting the big pharmas do what they are best at, commercializing products. The most elegant illustration of this approach is Isis, which declares itself as a “fully disintegrated” company that does not intend to be involved in late stage trials and marketing. Although Isis still hasn’t enjoyed meaningful sales of its products, the number of programs it has and its pipeline diversification are overwhelming.
Isis, a $1.5B company with thirteen drugs in the clinic and an expected operating loss of only $25 million in 2008, is also one of the few development stage companies that may reach profitability without actual royalties on sales.
5) Field Of Activity
Evaluating the field in which a company operates is somewhat trickier than the other criteria, as it is hard to define the different parameters and the balance between them. These parameters include validation and prior success rates of the mechanism of action and the target, the competitive landscape, entry barriers and intellectual property. Because a validated field usually comes with plenty of competition (from drugs in the market and in the clinic), it is very hard to find a platform company that is optimally positioned.
Companies with a good blend can typically be divided into pioneers like Isis and Alnylam, whose technologies are still not commercialized and “next-generation” companies such as Immunogen and Seattle Genetics (SGEN) that improve a validated class of drugs. Isis and Alnylam are high risk companies because of the lack of complete validation of their fields, but they have a considerable gap over the rest of the market. Immunogen’s and Seattle Genetics’ markets are more mature and proven, but the companies are facing more competition.
It is plain to see that the five criteria are intertwined in a way that forces companies to combine them. It is impossible to have multiple candidates in the clinic without multiple collaboration deals, which are hard to strike without a good platform that can produce multiple promising candidates. Partnership deals often lead to an increase in the attention a specific area receives, but such deals may very well be a result of a clinical validation by a single drug candidate, which in turn, may lead to the validation of an entire field.
Exelixis as a Platform Company
Exelixis is active in the ever growing market of kinase inhibitors (KIs) for the treatment of cancer, that is, drugs that block the activity of kinases in cancer cells. Cancer cells are often described as cells that are out of control: They proliferate quickly, ignore death signals, invade nearby tissues and eventually metastasize to distant organs. Disease onset and advancement are associated with processes such as cell growth, motility and blood-vessel formation, which are governed by a complex network made of kinases. Thus, blocking these processes by inhibiting the relevant kinases has emerged as one of the most attractive approaches to fighting cancer.
Together with monoclonal antibodies, kinase inhibitors represent a paradigm shift in cancer treatment from cytotoxic agents to targeted therapies, a trend that is constantly growing. Like antibodies for cancer, kinase inhibitors target tumors while sparing healthy cells and consequently lead to better activity with fewer side effects.
Kinase inhibitors, however, possess several advantages over antibodies. The most evident advantage is that KIs can hit targets inside the cell while antibodies can only bind targets presented on the cell surface, so internal targets are approachable only by KIs. Another advantage is the fact that KIs can be given orally, which is a major factor in terms of patient convenience, especially given the typical long treatment duration associated with targeted therapies. Another advantage, which will be later discussed in the article, is the ability to produce KIs that hit several targets at once.
The commercial potential of kinase inhibitors for cancer was validated by the approval of Novartis’ (NVS) Gleevec in 2001. Gleevec, which still remains the best selling kinase inhibitor drug, is considered one of the biggest success stories of modern medicine, owing to its long lasting benefit and excellent safety profile. Naturally, Gleevec’s success spurred considerable efforts to develop additional kinase inhibitors for cancer.
Seven years later, the field of kinase inhibitors is as active as ever, with eight additional approved drugs (three of which were launched only last year) and over a hundred kinase inhibitors in the clinic, most of which are evaluated for oncology indications. The implication of kinases in cancer is constantly being elucidated and today it is clear that they play a cardinal role in virtually every type of cancer. Commercial-wise, the market of kinase inhibitors is poised to grow at a fast pace thanks to new approvals as well as label extensions for marketed drugs.
It should be noted that although the benefit of kinase inhibitors is undeniable, Gleevec’s success in terms of duration of response and safety profile has not been replicated by other agents. In fact, many patients eventually develop resistance even against Gleevec.
The arms race between cancer and kinase inhibitors is just beginning, and the industry is placing its hopes on new approaches to developing better drugs. Some of these approaches include identifying new targets as well as new binding sites within known targets, simultaneously inhibiting a target with more than one drug, tailoring drugs for well defined populations and inhibiting several targets at once.
Examining Exelixis based on four of the five criteria - field of activity, a validated platform, multiple candidates and large partners makes it look very promising. The KI field is a highly validated field with plenty of growth ahead, so investing in this field seems like a smart thing to do.
With respect to clinical validation of the platform, several of Exelixis’ compounds have shown activity in humans. Although this activity has been shown in preliminary trials, it seems that Exelixis has one of the most promising pipelines in the industry. There are obviously no problems on the partnerships front, as Exelixis is involved with some high profile names from the likes of GlaxoSmithKline (GSK), Genentech (DNA), Bristol-Myers Squibb (BMY) and Wyeth (WYE). In terms of number of compounds in clinical trials, Exelixis has 11 drug candidates in over 20 clinical trials, which is quite impressive for a company its size.
The biggest issue with any KI company is the challenging competitive landscape. Because the field is so promising, it also became extremely crowded and competitive. There is no “top dog” in kinase inhibitors, but rather there are dozens of participants, from small biotechs to the world’s largest pharmaceutical companies, which naturally raises the question whether a single company can have a competitive edge over the rest of the market.
A company that develops kinase inhibitors typically does not have the luxury of blocking competitors by means of intellectual property, as all players are relatively free to operate in terms of the targets they explore, the way the compound binds the target, the approach for screening compounds and so on. Nevertheless, it would also be too simplistic to view this industry as a herd of “me too” companies, as there is still room for each company to differentiate itself.
Exelixis managed to differentiate itself based on three factors.
1) The Biggest Small Company
Exelixis is a small pure-play company focused entirely on kinase inhibitors. The risks of investing in small cap biotechs are evident, but so are the benefits. Exelixis provides 100% exposure to the field of kinase inhibitors, with a strong emphasis on cancer. This is something investors will not find in any of the larger players, no matter how active they are, not to mention the innovation and agility that are often absent in large pharmas.
For some reason, there is an inverse relationship between the innovation and the productivity of a company and its size. Smaller companies tend to be more productive in early stage drug development due to creative thinking and better maximization of resources. When it comes to the costly clinical stage, though, the large companies have the luxury of pursuing several paths in parallel as well as the capability of coping with the logistics and regulatory challenges.
One advantage Exelixis has over the myriad of small biotechs is the diversity and scope of its pipeline. As can be seen in the table below, in terms of the number of compounds in clinical development, Exelixis is in the big leagues.
Notably, a comparison based on number of drugs in the pipeline is flawed because it does not say anything about the quality of these drugs, nor does it specify the different trials with every compound. For example, Exelixis has one candidate (XL184) in phase III for a very small indication as well as in three earlier trials, while Pfizer’s phase III compounds (Sutent and axitinib) are being evaluated in tens of trials, including several phase III trials for very large indications.
In addition, being a small company, several of Exelixis’ compounds are partnered, as opposed to the large players who own the majority of their portfolios. Nevertheless, as one-dimensional as this comparison may be, it illustrates the fact that Exelixis’ pipeline is focused in terms of the therapeutic area, yet very diverse in terms of the number of compounds and the targets they hit. Besides, the impact of 15% of product sales (in the form of royalties) on a company like Exelixis is substantially stronger than that of 85% of sales on a large pharmaceutical company.
2) Discrete Patient Populations
It has been well recognized that classifying patients based on the origin of the tumor (i.e. pancreatic, breast cancer) ignores countless other important differences in the biology of the disease. Today, most cancer drugs are primarily given based on the site of tumor origin and consequently lead to a wide variability in response, both in terms of benefits and side effects. In order to optimally treat cancer, patients should be classified based on additional factors, with a strong emphasis on the mechanisms involved in their disease.
Distinguishing between the different subtypes is instrumental in the case of targeted therapies such as kinase inhibitors and monoclonal antibodies, because these agents usually affect one or a few targets that may not be relevant for all patients. Genentech’s Herceptin, for instance, has no effect on 70-80% of breast cancer patients, but leads to a strong benefit in the rest of the patients, whose tumors utilize Herceptin’s target (Her2) in order to grow and advance. Had Herceptin (over $5 billion in sales estimated this year) been evaluated in unselected breast cancer patients, it would probably have never reached the market due to lack of efficacy.
The commercial potential of niche markets is, by definition, more limited, but the strategy of addressing discrete patient populations holds several key advantages over starting with larger populations. First and foremost, focusing on small, well defined populations can lead to overwhelming results in terms of activity, and consequently increase the likelihood of approval. Moreover, it may provide early validation of the target or the compound’s activity in humans, which decreases the risk and renders the compound more attractive to potential partners.
Second, the development process involves smaller trials, so it is faster and cheaper than that of large indications. Third, niche markets are less crowded with competitors, and often ignored by the large pharmas. Fourth, although the addressable market is more modest, the drug can generate recurring revenues that are desperately needed by small companies. Fifth, assuming that the target population is well defined and highly responsive to the drug, revenues per patient are higher compared to the average in the industry, both due to longer treatment periods and patients’ and insurers’ willingness to pay more if they know there is a high likelihood of long-lasting benefit.
This has led many companies to realize that it might be better to focus on a subset of patients instead of targeting the whole market. Exelixis is spearheading the pursuit of personalized medicine by examining the pathways tumors “hijack” in order to grow, and identifying the right patient populations that are likely to benefit from a particular compound’s spectrum of activity.
The company’s compounds hit targets that may be relevant across a wide range of malignancies, representing very large target markets. Nevertheless, the company made a strategic decision to initially pursue niche markets with limited treatment options and use approvals in these small markets as a stepping stone for additional indications. Such niche markets might be either rare cancers or well defined subsets of patients within a large population of patients. This strategy is already starting to bear fruit, judging by results from two candidates: XL184 and XL880.
XL184 entered the clinic in 2005 and is currently being evaluated in three indications. Its shortest route to market is an ongoing phase III trial in 315 medullary thyroid cancer [MTC] patients, as it could hit the market as early as 2011. XL184 was active in all advanced MTC patients in a recently published trial, so chances for approval are high. Advanced MTC is considered a highly unmet medical need but the market size for this indication is very small (less than 1000 patients in the US and Europe every year). Thus, the overall potential for this indication amounts to several tens of millions annually, but it can serve as a proof of concept for the drug’s activity and safety profile, decreasing the overall risk in additional programs.
XL880 is currently being developed in collaboration with GSK. In a recently published phase II trial, XL880 showed impressive activity in patients with papillary renal cancer. These patients, which account for 8-10% of kidney cancer cases, represent an attractive niche because they seem to be less responsive to the approved targeted agents for kidney cancer. In the phase II trial, XL880 demonstrated impressive activity in papillary renal cancer patients with an exceptional duration of responses, which may lead to a registration trial already next year, only four years after entering the clinic.
The immediate target population for this indication is estimated at 10,000 patients in the United States and Europe. Importantly, because XL880’s benefit in the phase II trial lasted, on average, more than a year, the commercial opportunity can be as high as $400M annually.
3) Multi-Targeted Approach
The concept of hitting a combination of kinases simultaneously has been gaining momentum in the industry, given the complexity and multi-factorial nature of the cancer. In order to develop and advance, cancer cells must acquire multiple distinct capabilities such as the capability to proliferate, resist death signals, create new blood vessels and invade nearby tissues. Each capability can be governed by multiple pathways which work independently, in parallel, and through interconnections to promote cancer development. Therefore, by hitting multiple targets at once, a compound may employ more than one mechanism of action and increase spectrum of activity.
Multi-targeted kinase inhibitors can also target several points in the same pathway for achieving a more pronounced effect and delay the emergence of drug resistance. There may also be a commercial value in multi-targeted KIs because one drug can theoretically replace two or three single targeted agents, thereby lowering the cost of treatment.
In recent years, three drugs thought to inhibit multiple kinases entered the market (Sutent, Nexavar and Tykerb), and are seen as a validation of this approach. Interestingly, even kinase inhibitors which were developed as single kinase inhibitors, often have some degree of activity against additional kinases, whether clinically relevant or not. A good example for that might be Gleevec, which had started as a single kinase inhibitor, but then was reborn as an inhibitor of at least two other kinases. At the moment, Exelixis seems to favor the multi-kinase inhibitors approach, with 7 multi-targeted and 4 single-targeted kinase inhibitors in the clinic.
Although there is a wide consensus that simultaneous inhibition of multiple targets may optimize the use of targeted therapies, some question the benefit of multi-targeted inhibitors pointing at several issues. Manipulating several targets at once adds more complexity, on top of the intrinsic complexity of developing a drug against one target. More targets means more variables and uncertainty in terms of the choosing the right combination, the degree of inhibition of each target and the safety profile.
The promiscuous activity of multi-targeted kinase inhibitors often leads to increased toxicity, so these agents are not likely to have an ideal safety profile and consequently may not be given in sufficiently high doses. In addition, it is not always clear whether the clinical activity is due to inhibition of one or multiple targets, and the role each target plays. Most of Nexavar’s activity, for instance, is probably not the result of inhibition of the initial primary target for which the compound was developed, but due to a secondary target. As a result, there are some who believe that simultaneous inhibition of multiple targets should be done with several single-targeted agents rather than with one multi-targeted agent.
Like everything else in drug development, there is no clear solution for this debate, as there is probably more than one way of fighting cancer with kinase inhibitors. The right approach should be chosen on a case by case basis, depending on the specific drug, patient characteristics and the target itself.
Notwithstanding this debate, choosing the right targets is the first, and perhaps the most important step in the process. There is a tradeoff between the extent of clinical validation and the competitive landscape for each target.
Developing drugs that hit validated targets decreases the risk of failure on the one hand, but also involves fierce competition from approved and investigational agents. Examples for two highly validated targets may be EGFR (targeted by Tarceva and Iressa) and Bcr-Abl (targeted by Gleevec, Sprycel and Tasigna). Dethroning an approved drug is a challenging task, not to mention potential competition from compounds in development, so even if a company has a drug that works, it will not necessarily succeed in getting market share.
It is important to understand, though, that presence of approved agents does not necessarily hamper the potential of new compounds. Acknowledging that advanced cancer is almost impossible to cure, the industry is now talking about making it a chronic disease by using many lines of therapy. Kinase inhibitors’ inability to cure the disease and the emergence of resistance creates the need for developing second and third lines of therapy as well as combination treatments. There is, for instance, room for EGFR inhibitors that are active in Tarceva resistant patients or can be given in combination with Tarceva to improve activity and delay the onset of resistance.
Targets for which there are no marketed drugs involve more risk, but also represent the potential of opening up new markets with little or no competition. Exelixis decided to focus on kinases that are still not clinically validated, but such that have a clear role in cancer biology, backed up by a large body of scientific evidence. These targets include MET, PI3K, Hedgehog and MEK. Obviously, there are plenty of other compounds in development that target these kinases, and it is still premature to assess how Exelixis will fare with the competition, but thanks to its size and focus, it managed to be at the forefront of these exciting field.
Exelixis’s approach of targeting the right patient populations and the fact that some of its compounds inhibit multiple kinases may help it differentiate its compounds. In some cases, Exelixis further decreases the risk by developing two distinct compounds against the same target. For example, XL147 and XL765 both inhibit PI3K but the latter inhibits an additional kinase in the same signaling pathway - mTOR. On top of being an independent attempt of striking PI3K, each compound represents a different approach for inhibiting the same pathway in terms of the ideal number of targets to hit in this pathway. It is still unclear which approach is superior, and in which cases, so pursuing both routes in parallel sounds like a good idea.
Summary
Exelixis operates in one of the most promising yet competitive markets in the pharmaceutical industry, where there are always multiple candidates against any given target. The company is not nor will it ever be the undisputed leader of the KI market; however, it managed to distinguish itself as a hybrid between a small biotech and a large pharmaceutical company.
On the one hand, it is an extremely focused company with one of the leading R&D teams in the industry. On the other, it has a broad and diverse pipeline of promising compounds that may be applicable to a wide array of cancers. Most of these compounds will fail either due to poor performance or competing agents, but overall, Exelixis should have enough shots on goal in order to make it.
Disclosure: Author is long EXEL, ISIS, IMGN & SGEN.
***********
The cash issue is the most important factor in thenear term, no doubt. I will try to touch on that matter in the future, but that's an issue everybody including company's mangement is aware of. I expect they'll start to monetizing their portfolio in Q4, after the GSK decision(s). I hear they are very close to siging a deal for one of their PI3K compounds, which are not part the GSK collaboration.
mfg ipollit[/b]
http://seekingalpha.com/article/93716-exelixis-a-platform-co…
Exelixis: A Platform Company for Oncology
by: Ohad Hammer posted on: September 03, 2008 | about stocks: EXEL
The pharmaceutical industry is in the midst of a severe innovation crisis, where more R&D money results in less approved drugs. In parallel, sales of most traditional blockbusters are being cannibalized by generic competition, forcing the big pharmas to exploit new concepts and rush new drug candidates to their pipelines.
Nearly half of all drugs that are expected to enter the clinic in the coming years will be aimed at the oncology market. Consequently, early stage drug development for cancer is going to be the most active area in the industry, with an abundance of investment opportunities, particularly in small and medium companies.
Many investors prefer to stay away from oncology drug development, where success rates are at a worrying low, hovering around 5% compared to 10% for the whole industry. Nevertheless, among the hundreds of publicly traded companies which are engaged in oncology, there may be a group of companies which have better chances of succeeding in such a monumental task. These companies are what I like to call platform companies.
In this article, I will try to explain what makes a good platform company and why Exelixis (EXEL) can be regarded as a good (yet not a perfect) example. For the sake of clarification, I certainly do not claim that platform companies are the only investment-worthy biotech companies, however, when it comes to early stage drug development, platform companies are an excellent place to start.
In general, platform companies can be evaluated based on five criteria:
1) A validated platform
Having a validated drug discovery platform is the first and most important criterion for defining a good platform company. The platform is typically comprised of a combination of technology, experienced personnel and intellectual property that can generate a stream of drug candidates. Most importantly, investing should be done only after a product of the platform demonstrates activity in clinical trials. Having a clinically validated product is not a guarantee for future success of the platform nor does it mean that the specific agent will reach the market, but it does imply that one or more of the platform’s products stand a reasonable chance of becoming a commercial drug. A validated platform may increase overall success rates, yet the odds of a particular drug candidate to make it all the way to approval are still low.
It is hard to define exactly what “clinical validation” means, but there should be a clear response in the form of disease control, tumor shrinkage or side effects, preferably with more than one candidate, in patients with limited treatment options. A good example for a platform validation may be Immunogen’s (IMGN) antibody-drug conjugation platform, which empowers antibodies by conjugating them to toxic drug payloads. T-DM1 is comprised of the blockbuster antibody, Herceptin, empowered by Immunogen’s technology. It had remarkable activity in two clinical trials in patients who had progressed while receiving Herceptin, and therefore can be regarded as a good clinical validation of Immunogen’s technology.
2) Specialization and Differentiation
The field of drug development is becoming more rich and versatile on the one hand, but also more complex and competitive on the other. Instead of relying on traditional trial and error and serendipity for discovering drugs, the industry is now moving to rational design and computerized tools, which require a great deal of expertise. This is why small companies must not spread their resources too thinly but instead focus on one field. By specializing in a certain area, and developing a deep understanding of the underlying science that stands at the basis of development, even a small company can achieve a competitive edge over the rest of the market.
The ability to block new entrants to the field by means of intellectual property is always an advantage, as clearly demonstrated in the case of Alnylam (ALNY) and Isis Pharmaceuticals (ISIS), which became the gate-keepers of their fields with the help of a draconian patent portfolio. Timing is also a crucial issue in finding a niche to specialize in, and since the process of developing a drug is a linear and lengthy one, a several years head start is needed in most cases.
3) Multiple shots on goal
A good platform company always has multiple agents in active development simultaneously, preferably from the same validated platform. No matter how promising a particular compound looks, it is imperative to remember that most drugs fail, including those that demonstrate spectacular results in early clinical trials; thus, banking on one or two early stage compounds is statistically futile.
Just to make it clear, active development does not include just having the compounds listed on the pipeline chart, waiting to fill the gap created by discontinuation of programs, but a constant pursuit of the compound’s development with the ultimate goal of advancing it into multiple clinical trials. Due to the enormous costs associated with drug development, small and medium companies cannot finance more than a handful of programs, which leads us to the next point.
4) Deep Pocketed Partners
Partnership deals with large pharmaceutical companies are the only viable options for a small company to obtain a broad pipeline. Collaborating with large partners serves two prime purposes. First, it allows small players to survive the time needed to develop drugs without infinite dilution. In addition, any partnership deal is perceived as a vote of confidence for the technology from a savvy player in the industry, which bodes well for investors’ confidence and valuation.
The dark side of partnership deals is that if the drug hits the market, the large partner enjoys most of the gains, but in most cases the risks and dilution accompanied with developing the drug independently are far graver. Trying to keep all the promising candidates in-house can turn these assets into a huge liability.
Small biotechs must realize what they are good at, feeding the industry with a large number of candidates, while letting the big pharmas do what they are best at, commercializing products. The most elegant illustration of this approach is Isis, which declares itself as a “fully disintegrated” company that does not intend to be involved in late stage trials and marketing. Although Isis still hasn’t enjoyed meaningful sales of its products, the number of programs it has and its pipeline diversification are overwhelming.
Isis, a $1.5B company with thirteen drugs in the clinic and an expected operating loss of only $25 million in 2008, is also one of the few development stage companies that may reach profitability without actual royalties on sales.
5) Field Of Activity
Evaluating the field in which a company operates is somewhat trickier than the other criteria, as it is hard to define the different parameters and the balance between them. These parameters include validation and prior success rates of the mechanism of action and the target, the competitive landscape, entry barriers and intellectual property. Because a validated field usually comes with plenty of competition (from drugs in the market and in the clinic), it is very hard to find a platform company that is optimally positioned.
Companies with a good blend can typically be divided into pioneers like Isis and Alnylam, whose technologies are still not commercialized and “next-generation” companies such as Immunogen and Seattle Genetics (SGEN) that improve a validated class of drugs. Isis and Alnylam are high risk companies because of the lack of complete validation of their fields, but they have a considerable gap over the rest of the market. Immunogen’s and Seattle Genetics’ markets are more mature and proven, but the companies are facing more competition.
It is plain to see that the five criteria are intertwined in a way that forces companies to combine them. It is impossible to have multiple candidates in the clinic without multiple collaboration deals, which are hard to strike without a good platform that can produce multiple promising candidates. Partnership deals often lead to an increase in the attention a specific area receives, but such deals may very well be a result of a clinical validation by a single drug candidate, which in turn, may lead to the validation of an entire field.
Exelixis as a Platform Company
Exelixis is active in the ever growing market of kinase inhibitors (KIs) for the treatment of cancer, that is, drugs that block the activity of kinases in cancer cells. Cancer cells are often described as cells that are out of control: They proliferate quickly, ignore death signals, invade nearby tissues and eventually metastasize to distant organs. Disease onset and advancement are associated with processes such as cell growth, motility and blood-vessel formation, which are governed by a complex network made of kinases. Thus, blocking these processes by inhibiting the relevant kinases has emerged as one of the most attractive approaches to fighting cancer.
Together with monoclonal antibodies, kinase inhibitors represent a paradigm shift in cancer treatment from cytotoxic agents to targeted therapies, a trend that is constantly growing. Like antibodies for cancer, kinase inhibitors target tumors while sparing healthy cells and consequently lead to better activity with fewer side effects.
Kinase inhibitors, however, possess several advantages over antibodies. The most evident advantage is that KIs can hit targets inside the cell while antibodies can only bind targets presented on the cell surface, so internal targets are approachable only by KIs. Another advantage is the fact that KIs can be given orally, which is a major factor in terms of patient convenience, especially given the typical long treatment duration associated with targeted therapies. Another advantage, which will be later discussed in the article, is the ability to produce KIs that hit several targets at once.
The commercial potential of kinase inhibitors for cancer was validated by the approval of Novartis’ (NVS) Gleevec in 2001. Gleevec, which still remains the best selling kinase inhibitor drug, is considered one of the biggest success stories of modern medicine, owing to its long lasting benefit and excellent safety profile. Naturally, Gleevec’s success spurred considerable efforts to develop additional kinase inhibitors for cancer.
Seven years later, the field of kinase inhibitors is as active as ever, with eight additional approved drugs (three of which were launched only last year) and over a hundred kinase inhibitors in the clinic, most of which are evaluated for oncology indications. The implication of kinases in cancer is constantly being elucidated and today it is clear that they play a cardinal role in virtually every type of cancer. Commercial-wise, the market of kinase inhibitors is poised to grow at a fast pace thanks to new approvals as well as label extensions for marketed drugs.
It should be noted that although the benefit of kinase inhibitors is undeniable, Gleevec’s success in terms of duration of response and safety profile has not been replicated by other agents. In fact, many patients eventually develop resistance even against Gleevec.
The arms race between cancer and kinase inhibitors is just beginning, and the industry is placing its hopes on new approaches to developing better drugs. Some of these approaches include identifying new targets as well as new binding sites within known targets, simultaneously inhibiting a target with more than one drug, tailoring drugs for well defined populations and inhibiting several targets at once.
Examining Exelixis based on four of the five criteria - field of activity, a validated platform, multiple candidates and large partners makes it look very promising. The KI field is a highly validated field with plenty of growth ahead, so investing in this field seems like a smart thing to do.
With respect to clinical validation of the platform, several of Exelixis’ compounds have shown activity in humans. Although this activity has been shown in preliminary trials, it seems that Exelixis has one of the most promising pipelines in the industry. There are obviously no problems on the partnerships front, as Exelixis is involved with some high profile names from the likes of GlaxoSmithKline (GSK), Genentech (DNA), Bristol-Myers Squibb (BMY) and Wyeth (WYE). In terms of number of compounds in clinical trials, Exelixis has 11 drug candidates in over 20 clinical trials, which is quite impressive for a company its size.
The biggest issue with any KI company is the challenging competitive landscape. Because the field is so promising, it also became extremely crowded and competitive. There is no “top dog” in kinase inhibitors, but rather there are dozens of participants, from small biotechs to the world’s largest pharmaceutical companies, which naturally raises the question whether a single company can have a competitive edge over the rest of the market.
A company that develops kinase inhibitors typically does not have the luxury of blocking competitors by means of intellectual property, as all players are relatively free to operate in terms of the targets they explore, the way the compound binds the target, the approach for screening compounds and so on. Nevertheless, it would also be too simplistic to view this industry as a herd of “me too” companies, as there is still room for each company to differentiate itself.
Exelixis managed to differentiate itself based on three factors.
1) The Biggest Small Company
Exelixis is a small pure-play company focused entirely on kinase inhibitors. The risks of investing in small cap biotechs are evident, but so are the benefits. Exelixis provides 100% exposure to the field of kinase inhibitors, with a strong emphasis on cancer. This is something investors will not find in any of the larger players, no matter how active they are, not to mention the innovation and agility that are often absent in large pharmas.
For some reason, there is an inverse relationship between the innovation and the productivity of a company and its size. Smaller companies tend to be more productive in early stage drug development due to creative thinking and better maximization of resources. When it comes to the costly clinical stage, though, the large companies have the luxury of pursuing several paths in parallel as well as the capability of coping with the logistics and regulatory challenges.
One advantage Exelixis has over the myriad of small biotechs is the diversity and scope of its pipeline. As can be seen in the table below, in terms of the number of compounds in clinical development, Exelixis is in the big leagues.
Notably, a comparison based on number of drugs in the pipeline is flawed because it does not say anything about the quality of these drugs, nor does it specify the different trials with every compound. For example, Exelixis has one candidate (XL184) in phase III for a very small indication as well as in three earlier trials, while Pfizer’s phase III compounds (Sutent and axitinib) are being evaluated in tens of trials, including several phase III trials for very large indications.
In addition, being a small company, several of Exelixis’ compounds are partnered, as opposed to the large players who own the majority of their portfolios. Nevertheless, as one-dimensional as this comparison may be, it illustrates the fact that Exelixis’ pipeline is focused in terms of the therapeutic area, yet very diverse in terms of the number of compounds and the targets they hit. Besides, the impact of 15% of product sales (in the form of royalties) on a company like Exelixis is substantially stronger than that of 85% of sales on a large pharmaceutical company.
2) Discrete Patient Populations
It has been well recognized that classifying patients based on the origin of the tumor (i.e. pancreatic, breast cancer) ignores countless other important differences in the biology of the disease. Today, most cancer drugs are primarily given based on the site of tumor origin and consequently lead to a wide variability in response, both in terms of benefits and side effects. In order to optimally treat cancer, patients should be classified based on additional factors, with a strong emphasis on the mechanisms involved in their disease.
Distinguishing between the different subtypes is instrumental in the case of targeted therapies such as kinase inhibitors and monoclonal antibodies, because these agents usually affect one or a few targets that may not be relevant for all patients. Genentech’s Herceptin, for instance, has no effect on 70-80% of breast cancer patients, but leads to a strong benefit in the rest of the patients, whose tumors utilize Herceptin’s target (Her2) in order to grow and advance. Had Herceptin (over $5 billion in sales estimated this year) been evaluated in unselected breast cancer patients, it would probably have never reached the market due to lack of efficacy.
The commercial potential of niche markets is, by definition, more limited, but the strategy of addressing discrete patient populations holds several key advantages over starting with larger populations. First and foremost, focusing on small, well defined populations can lead to overwhelming results in terms of activity, and consequently increase the likelihood of approval. Moreover, it may provide early validation of the target or the compound’s activity in humans, which decreases the risk and renders the compound more attractive to potential partners.
Second, the development process involves smaller trials, so it is faster and cheaper than that of large indications. Third, niche markets are less crowded with competitors, and often ignored by the large pharmas. Fourth, although the addressable market is more modest, the drug can generate recurring revenues that are desperately needed by small companies. Fifth, assuming that the target population is well defined and highly responsive to the drug, revenues per patient are higher compared to the average in the industry, both due to longer treatment periods and patients’ and insurers’ willingness to pay more if they know there is a high likelihood of long-lasting benefit.
This has led many companies to realize that it might be better to focus on a subset of patients instead of targeting the whole market. Exelixis is spearheading the pursuit of personalized medicine by examining the pathways tumors “hijack” in order to grow, and identifying the right patient populations that are likely to benefit from a particular compound’s spectrum of activity.
The company’s compounds hit targets that may be relevant across a wide range of malignancies, representing very large target markets. Nevertheless, the company made a strategic decision to initially pursue niche markets with limited treatment options and use approvals in these small markets as a stepping stone for additional indications. Such niche markets might be either rare cancers or well defined subsets of patients within a large population of patients. This strategy is already starting to bear fruit, judging by results from two candidates: XL184 and XL880.
XL184 entered the clinic in 2005 and is currently being evaluated in three indications. Its shortest route to market is an ongoing phase III trial in 315 medullary thyroid cancer [MTC] patients, as it could hit the market as early as 2011. XL184 was active in all advanced MTC patients in a recently published trial, so chances for approval are high. Advanced MTC is considered a highly unmet medical need but the market size for this indication is very small (less than 1000 patients in the US and Europe every year). Thus, the overall potential for this indication amounts to several tens of millions annually, but it can serve as a proof of concept for the drug’s activity and safety profile, decreasing the overall risk in additional programs.
XL880 is currently being developed in collaboration with GSK. In a recently published phase II trial, XL880 showed impressive activity in patients with papillary renal cancer. These patients, which account for 8-10% of kidney cancer cases, represent an attractive niche because they seem to be less responsive to the approved targeted agents for kidney cancer. In the phase II trial, XL880 demonstrated impressive activity in papillary renal cancer patients with an exceptional duration of responses, which may lead to a registration trial already next year, only four years after entering the clinic.
The immediate target population for this indication is estimated at 10,000 patients in the United States and Europe. Importantly, because XL880’s benefit in the phase II trial lasted, on average, more than a year, the commercial opportunity can be as high as $400M annually.
3) Multi-Targeted Approach
The concept of hitting a combination of kinases simultaneously has been gaining momentum in the industry, given the complexity and multi-factorial nature of the cancer. In order to develop and advance, cancer cells must acquire multiple distinct capabilities such as the capability to proliferate, resist death signals, create new blood vessels and invade nearby tissues. Each capability can be governed by multiple pathways which work independently, in parallel, and through interconnections to promote cancer development. Therefore, by hitting multiple targets at once, a compound may employ more than one mechanism of action and increase spectrum of activity.
Multi-targeted kinase inhibitors can also target several points in the same pathway for achieving a more pronounced effect and delay the emergence of drug resistance. There may also be a commercial value in multi-targeted KIs because one drug can theoretically replace two or three single targeted agents, thereby lowering the cost of treatment.
In recent years, three drugs thought to inhibit multiple kinases entered the market (Sutent, Nexavar and Tykerb), and are seen as a validation of this approach. Interestingly, even kinase inhibitors which were developed as single kinase inhibitors, often have some degree of activity against additional kinases, whether clinically relevant or not. A good example for that might be Gleevec, which had started as a single kinase inhibitor, but then was reborn as an inhibitor of at least two other kinases. At the moment, Exelixis seems to favor the multi-kinase inhibitors approach, with 7 multi-targeted and 4 single-targeted kinase inhibitors in the clinic.
Although there is a wide consensus that simultaneous inhibition of multiple targets may optimize the use of targeted therapies, some question the benefit of multi-targeted inhibitors pointing at several issues. Manipulating several targets at once adds more complexity, on top of the intrinsic complexity of developing a drug against one target. More targets means more variables and uncertainty in terms of the choosing the right combination, the degree of inhibition of each target and the safety profile.
The promiscuous activity of multi-targeted kinase inhibitors often leads to increased toxicity, so these agents are not likely to have an ideal safety profile and consequently may not be given in sufficiently high doses. In addition, it is not always clear whether the clinical activity is due to inhibition of one or multiple targets, and the role each target plays. Most of Nexavar’s activity, for instance, is probably not the result of inhibition of the initial primary target for which the compound was developed, but due to a secondary target. As a result, there are some who believe that simultaneous inhibition of multiple targets should be done with several single-targeted agents rather than with one multi-targeted agent.
Like everything else in drug development, there is no clear solution for this debate, as there is probably more than one way of fighting cancer with kinase inhibitors. The right approach should be chosen on a case by case basis, depending on the specific drug, patient characteristics and the target itself.
Notwithstanding this debate, choosing the right targets is the first, and perhaps the most important step in the process. There is a tradeoff between the extent of clinical validation and the competitive landscape for each target.
Developing drugs that hit validated targets decreases the risk of failure on the one hand, but also involves fierce competition from approved and investigational agents. Examples for two highly validated targets may be EGFR (targeted by Tarceva and Iressa) and Bcr-Abl (targeted by Gleevec, Sprycel and Tasigna). Dethroning an approved drug is a challenging task, not to mention potential competition from compounds in development, so even if a company has a drug that works, it will not necessarily succeed in getting market share.
It is important to understand, though, that presence of approved agents does not necessarily hamper the potential of new compounds. Acknowledging that advanced cancer is almost impossible to cure, the industry is now talking about making it a chronic disease by using many lines of therapy. Kinase inhibitors’ inability to cure the disease and the emergence of resistance creates the need for developing second and third lines of therapy as well as combination treatments. There is, for instance, room for EGFR inhibitors that are active in Tarceva resistant patients or can be given in combination with Tarceva to improve activity and delay the onset of resistance.
Targets for which there are no marketed drugs involve more risk, but also represent the potential of opening up new markets with little or no competition. Exelixis decided to focus on kinases that are still not clinically validated, but such that have a clear role in cancer biology, backed up by a large body of scientific evidence. These targets include MET, PI3K, Hedgehog and MEK. Obviously, there are plenty of other compounds in development that target these kinases, and it is still premature to assess how Exelixis will fare with the competition, but thanks to its size and focus, it managed to be at the forefront of these exciting field.
Exelixis’s approach of targeting the right patient populations and the fact that some of its compounds inhibit multiple kinases may help it differentiate its compounds. In some cases, Exelixis further decreases the risk by developing two distinct compounds against the same target. For example, XL147 and XL765 both inhibit PI3K but the latter inhibits an additional kinase in the same signaling pathway - mTOR. On top of being an independent attempt of striking PI3K, each compound represents a different approach for inhibiting the same pathway in terms of the ideal number of targets to hit in this pathway. It is still unclear which approach is superior, and in which cases, so pursuing both routes in parallel sounds like a good idea.
Summary
Exelixis operates in one of the most promising yet competitive markets in the pharmaceutical industry, where there are always multiple candidates against any given target. The company is not nor will it ever be the undisputed leader of the KI market; however, it managed to distinguish itself as a hybrid between a small biotech and a large pharmaceutical company.
On the one hand, it is an extremely focused company with one of the leading R&D teams in the industry. On the other, it has a broad and diverse pipeline of promising compounds that may be applicable to a wide array of cancers. Most of these compounds will fail either due to poor performance or competing agents, but overall, Exelixis should have enough shots on goal in order to make it.
Disclosure: Author is long EXEL, ISIS, IMGN & SGEN.
***********
The cash issue is the most important factor in thenear term, no doubt. I will try to touch on that matter in the future, but that's an issue everybody including company's mangement is aware of. I expect they'll start to monetizing their portfolio in Q4, after the GSK decision(s). I hear they are very close to siging a deal for one of their PI3K compounds, which are not part the GSK collaboration.
mfg ipollit" target="_blank" rel="nofollow ugc noopener">http://seekingalpha.com/article/93716-exelixis-a-platform-co…
Exelixis: A Platform Company for Oncology
by: Ohad Hammer posted on: September 03, 2008 | about stocks: EXEL
The pharmaceutical industry is in the midst of a severe innovation crisis, where more R&D money results in less approved drugs. In parallel, sales of most traditional blockbusters are being cannibalized by generic competition, forcing the big pharmas to exploit new concepts and rush new drug candidates to their pipelines.
Nearly half of all drugs that are expected to enter the clinic in the coming years will be aimed at the oncology market. Consequently, early stage drug development for cancer is going to be the most active area in the industry, with an abundance of investment opportunities, particularly in small and medium companies.
Many investors prefer to stay away from oncology drug development, where success rates are at a worrying low, hovering around 5% compared to 10% for the whole industry. Nevertheless, among the hundreds of publicly traded companies which are engaged in oncology, there may be a group of companies which have better chances of succeeding in such a monumental task. These companies are what I like to call platform companies.
In this article, I will try to explain what makes a good platform company and why Exelixis (EXEL) can be regarded as a good (yet not a perfect) example. For the sake of clarification, I certainly do not claim that platform companies are the only investment-worthy biotech companies, however, when it comes to early stage drug development, platform companies are an excellent place to start.
In general, platform companies can be evaluated based on five criteria:
1) A validated platform
Having a validated drug discovery platform is the first and most important criterion for defining a good platform company. The platform is typically comprised of a combination of technology, experienced personnel and intellectual property that can generate a stream of drug candidates. Most importantly, investing should be done only after a product of the platform demonstrates activity in clinical trials. Having a clinically validated product is not a guarantee for future success of the platform nor does it mean that the specific agent will reach the market, but it does imply that one or more of the platform’s products stand a reasonable chance of becoming a commercial drug. A validated platform may increase overall success rates, yet the odds of a particular drug candidate to make it all the way to approval are still low.
It is hard to define exactly what “clinical validation” means, but there should be a clear response in the form of disease control, tumor shrinkage or side effects, preferably with more than one candidate, in patients with limited treatment options. A good example for a platform validation may be Immunogen’s (IMGN) antibody-drug conjugation platform, which empowers antibodies by conjugating them to toxic drug payloads. T-DM1 is comprised of the blockbuster antibody, Herceptin, empowered by Immunogen’s technology. It had remarkable activity in two clinical trials in patients who had progressed while receiving Herceptin, and therefore can be regarded as a good clinical validation of Immunogen’s technology.
2) Specialization and Differentiation
The field of drug development is becoming more rich and versatile on the one hand, but also more complex and competitive on the other. Instead of relying on traditional trial and error and serendipity for discovering drugs, the industry is now moving to rational design and computerized tools, which require a great deal of expertise. This is why small companies must not spread their resources too thinly but instead focus on one field. By specializing in a certain area, and developing a deep understanding of the underlying science that stands at the basis of development, even a small company can achieve a competitive edge over the rest of the market.
The ability to block new entrants to the field by means of intellectual property is always an advantage, as clearly demonstrated in the case of Alnylam (ALNY) and Isis Pharmaceuticals (ISIS), which became the gate-keepers of their fields with the help of a draconian patent portfolio. Timing is also a crucial issue in finding a niche to specialize in, and since the process of developing a drug is a linear and lengthy one, a several years head start is needed in most cases.
3) Multiple shots on goal
A good platform company always has multiple agents in active development simultaneously, preferably from the same validated platform. No matter how promising a particular compound looks, it is imperative to remember that most drugs fail, including those that demonstrate spectacular results in early clinical trials; thus, banking on one or two early stage compounds is statistically futile.
Just to make it clear, active development does not include just having the compounds listed on the pipeline chart, waiting to fill the gap created by discontinuation of programs, but a constant pursuit of the compound’s development with the ultimate goal of advancing it into multiple clinical trials. Due to the enormous costs associated with drug development, small and medium companies cannot finance more than a handful of programs, which leads us to the next point.
4) Deep Pocketed Partners
Partnership deals with large pharmaceutical companies are the only viable options for a small company to obtain a broad pipeline. Collaborating with large partners serves two prime purposes. First, it allows small players to survive the time needed to develop drugs without infinite dilution. In addition, any partnership deal is perceived as a vote of confidence for the technology from a savvy player in the industry, which bodes well for investors’ confidence and valuation.
The dark side of partnership deals is that if the drug hits the market, the large partner enjoys most of the gains, but in most cases the risks and dilution accompanied with developing the drug independently are far graver. Trying to keep all the promising candidates in-house can turn these assets into a huge liability.
Small biotechs must realize what they are good at, feeding the industry with a large number of candidates, while letting the big pharmas do what they are best at, commercializing products. The most elegant illustration of this approach is Isis, which declares itself as a “fully disintegrated” company that does not intend to be involved in late stage trials and marketing. Although Isis still hasn’t enjoyed meaningful sales of its products, the number of programs it has and its pipeline diversification are overwhelming.
Isis, a $1.5B company with thirteen drugs in the clinic and an expected operating loss of only $25 million in 2008, is also one of the few development stage companies that may reach profitability without actual royalties on sales.
5) Field Of Activity
Evaluating the field in which a company operates is somewhat trickier than the other criteria, as it is hard to define the different parameters and the balance between them. These parameters include validation and prior success rates of the mechanism of action and the target, the competitive landscape, entry barriers and intellectual property. Because a validated field usually comes with plenty of competition (from drugs in the market and in the clinic), it is very hard to find a platform company that is optimally positioned.
Companies with a good blend can typically be divided into pioneers like Isis and Alnylam, whose technologies are still not commercialized and “next-generation” companies such as Immunogen and Seattle Genetics (SGEN) that improve a validated class of drugs. Isis and Alnylam are high risk companies because of the lack of complete validation of their fields, but they have a considerable gap over the rest of the market. Immunogen’s and Seattle Genetics’ markets are more mature and proven, but the companies are facing more competition.
It is plain to see that the five criteria are intertwined in a way that forces companies to combine them. It is impossible to have multiple candidates in the clinic without multiple collaboration deals, which are hard to strike without a good platform that can produce multiple promising candidates. Partnership deals often lead to an increase in the attention a specific area receives, but such deals may very well be a result of a clinical validation by a single drug candidate, which in turn, may lead to the validation of an entire field.
Exelixis as a Platform Company
Exelixis is active in the ever growing market of kinase inhibitors (KIs) for the treatment of cancer, that is, drugs that block the activity of kinases in cancer cells. Cancer cells are often described as cells that are out of control: They proliferate quickly, ignore death signals, invade nearby tissues and eventually metastasize to distant organs. Disease onset and advancement are associated with processes such as cell growth, motility and blood-vessel formation, which are governed by a complex network made of kinases. Thus, blocking these processes by inhibiting the relevant kinases has emerged as one of the most attractive approaches to fighting cancer.
Together with monoclonal antibodies, kinase inhibitors represent a paradigm shift in cancer treatment from cytotoxic agents to targeted therapies, a trend that is constantly growing. Like antibodies for cancer, kinase inhibitors target tumors while sparing healthy cells and consequently lead to better activity with fewer side effects.
Kinase inhibitors, however, possess several advantages over antibodies. The most evident advantage is that KIs can hit targets inside the cell while antibodies can only bind targets presented on the cell surface, so internal targets are approachable only by KIs. Another advantage is the fact that KIs can be given orally, which is a major factor in terms of patient convenience, especially given the typical long treatment duration associated with targeted therapies. Another advantage, which will be later discussed in the article, is the ability to produce KIs that hit several targets at once.
The commercial potential of kinase inhibitors for cancer was validated by the approval of Novartis’ (NVS) Gleevec in 2001. Gleevec, which still remains the best selling kinase inhibitor drug, is considered one of the biggest success stories of modern medicine, owing to its long lasting benefit and excellent safety profile. Naturally, Gleevec’s success spurred considerable efforts to develop additional kinase inhibitors for cancer.
Seven years later, the field of kinase inhibitors is as active as ever, with eight additional approved drugs (three of which were launched only last year) and over a hundred kinase inhibitors in the clinic, most of which are evaluated for oncology indications. The implication of kinases in cancer is constantly being elucidated and today it is clear that they play a cardinal role in virtually every type of cancer. Commercial-wise, the market of kinase inhibitors is poised to grow at a fast pace thanks to new approvals as well as label extensions for marketed drugs.
It should be noted that although the benefit of kinase inhibitors is undeniable, Gleevec’s success in terms of duration of response and safety profile has not been replicated by other agents. In fact, many patients eventually develop resistance even against Gleevec.
The arms race between cancer and kinase inhibitors is just beginning, and the industry is placing its hopes on new approaches to developing better drugs. Some of these approaches include identifying new targets as well as new binding sites within known targets, simultaneously inhibiting a target with more than one drug, tailoring drugs for well defined populations and inhibiting several targets at once.
Examining Exelixis based on four of the five criteria - field of activity, a validated platform, multiple candidates and large partners makes it look very promising. The KI field is a highly validated field with plenty of growth ahead, so investing in this field seems like a smart thing to do.
With respect to clinical validation of the platform, several of Exelixis’ compounds have shown activity in humans. Although this activity has been shown in preliminary trials, it seems that Exelixis has one of the most promising pipelines in the industry. There are obviously no problems on the partnerships front, as Exelixis is involved with some high profile names from the likes of GlaxoSmithKline (GSK), Genentech (DNA), Bristol-Myers Squibb (BMY) and Wyeth (WYE). In terms of number of compounds in clinical trials, Exelixis has 11 drug candidates in over 20 clinical trials, which is quite impressive for a company its size.
The biggest issue with any KI company is the challenging competitive landscape. Because the field is so promising, it also became extremely crowded and competitive. There is no “top dog” in kinase inhibitors, but rather there are dozens of participants, from small biotechs to the world’s largest pharmaceutical companies, which naturally raises the question whether a single company can have a competitive edge over the rest of the market.
A company that develops kinase inhibitors typically does not have the luxury of blocking competitors by means of intellectual property, as all players are relatively free to operate in terms of the targets they explore, the way the compound binds the target, the approach for screening compounds and so on. Nevertheless, it would also be too simplistic to view this industry as a herd of “me too” companies, as there is still room for each company to differentiate itself.
Exelixis managed to differentiate itself based on three factors.
1) The Biggest Small Company
Exelixis is a small pure-play company focused entirely on kinase inhibitors. The risks of investing in small cap biotechs are evident, but so are the benefits. Exelixis provides 100% exposure to the field of kinase inhibitors, with a strong emphasis on cancer. This is something investors will not find in any of the larger players, no matter how active they are, not to mention the innovation and agility that are often absent in large pharmas.
For some reason, there is an inverse relationship between the innovation and the productivity of a company and its size. Smaller companies tend to be more productive in early stage drug development due to creative thinking and better maximization of resources. When it comes to the costly clinical stage, though, the large companies have the luxury of pursuing several paths in parallel as well as the capability of coping with the logistics and regulatory challenges.
One advantage Exelixis has over the myriad of small biotechs is the diversity and scope of its pipeline. As can be seen in the table below, in terms of the number of compounds in clinical development, Exelixis is in the big leagues.
Notably, a comparison based on number of drugs in the pipeline is flawed because it does not say anything about the quality of these drugs, nor does it specify the different trials with every compound. For example, Exelixis has one candidate (XL184) in phase III for a very small indication as well as in three earlier trials, while Pfizer’s phase III compounds (Sutent and axitinib) are being evaluated in tens of trials, including several phase III trials for very large indications.
In addition, being a small company, several of Exelixis’ compounds are partnered, as opposed to the large players who own the majority of their portfolios. Nevertheless, as one-dimensional as this comparison may be, it illustrates the fact that Exelixis’ pipeline is focused in terms of the therapeutic area, yet very diverse in terms of the number of compounds and the targets they hit. Besides, the impact of 15% of product sales (in the form of royalties) on a company like Exelixis is substantially stronger than that of 85% of sales on a large pharmaceutical company.
2) Discrete Patient Populations
It has been well recognized that classifying patients based on the origin of the tumor (i.e. pancreatic, breast cancer) ignores countless other important differences in the biology of the disease. Today, most cancer drugs are primarily given based on the site of tumor origin and consequently lead to a wide variability in response, both in terms of benefits and side effects. In order to optimally treat cancer, patients should be classified based on additional factors, with a strong emphasis on the mechanisms involved in their disease.
Distinguishing between the different subtypes is instrumental in the case of targeted therapies such as kinase inhibitors and monoclonal antibodies, because these agents usually affect one or a few targets that may not be relevant for all patients. Genentech’s Herceptin, for instance, has no effect on 70-80% of breast cancer patients, but leads to a strong benefit in the rest of the patients, whose tumors utilize Herceptin’s target (Her2) in order to grow and advance. Had Herceptin (over $5 billion in sales estimated this year) been evaluated in unselected breast cancer patients, it would probably have never reached the market due to lack of efficacy.
The commercial potential of niche markets is, by definition, more limited, but the strategy of addressing discrete patient populations holds several key advantages over starting with larger populations. First and foremost, focusing on small, well defined populations can lead to overwhelming results in terms of activity, and consequently increase the likelihood of approval. Moreover, it may provide early validation of the target or the compound’s activity in humans, which decreases the risk and renders the compound more attractive to potential partners.
Second, the development process involves smaller trials, so it is faster and cheaper than that of large indications. Third, niche markets are less crowded with competitors, and often ignored by the large pharmas. Fourth, although the addressable market is more modest, the drug can generate recurring revenues that are desperately needed by small companies. Fifth, assuming that the target population is well defined and highly responsive to the drug, revenues per patient are higher compared to the average in the industry, both due to longer treatment periods and patients’ and insurers’ willingness to pay more if they know there is a high likelihood of long-lasting benefit.
This has led many companies to realize that it might be better to focus on a subset of patients instead of targeting the whole market. Exelixis is spearheading the pursuit of personalized medicine by examining the pathways tumors “hijack” in order to grow, and identifying the right patient populations that are likely to benefit from a particular compound’s spectrum of activity.
The company’s compounds hit targets that may be relevant across a wide range of malignancies, representing very large target markets. Nevertheless, the company made a strategic decision to initially pursue niche markets with limited treatment options and use approvals in these small markets as a stepping stone for additional indications. Such niche markets might be either rare cancers or well defined subsets of patients within a large population of patients. This strategy is already starting to bear fruit, judging by results from two candidates: XL184 and XL880.
XL184 entered the clinic in 2005 and is currently being evaluated in three indications. Its shortest route to market is an ongoing phase III trial in 315 medullary thyroid cancer [MTC] patients, as it could hit the market as early as 2011. XL184 was active in all advanced MTC patients in a recently published trial, so chances for approval are high. Advanced MTC is considered a highly unmet medical need but the market size for this indication is very small (less than 1000 patients in the US and Europe every year). Thus, the overall potential for this indication amounts to several tens of millions annually, but it can serve as a proof of concept for the drug’s activity and safety profile, decreasing the overall risk in additional programs.
XL880 is currently being developed in collaboration with GSK. In a recently published phase II trial, XL880 showed impressive activity in patients with papillary renal cancer. These patients, which account for 8-10% of kidney cancer cases, represent an attractive niche because they seem to be less responsive to the approved targeted agents for kidney cancer. In the phase II trial, XL880 demonstrated impressive activity in papillary renal cancer patients with an exceptional duration of responses, which may lead to a registration trial already next year, only four years after entering the clinic.
The immediate target population for this indication is estimated at 10,000 patients in the United States and Europe. Importantly, because XL880’s benefit in the phase II trial lasted, on average, more than a year, the commercial opportunity can be as high as $400M annually.
3) Multi-Targeted Approach
The concept of hitting a combination of kinases simultaneously has been gaining momentum in the industry, given the complexity and multi-factorial nature of the cancer. In order to develop and advance, cancer cells must acquire multiple distinct capabilities such as the capability to proliferate, resist death signals, create new blood vessels and invade nearby tissues. Each capability can be governed by multiple pathways which work independently, in parallel, and through interconnections to promote cancer development. Therefore, by hitting multiple targets at once, a compound may employ more than one mechanism of action and increase spectrum of activity.
Multi-targeted kinase inhibitors can also target several points in the same pathway for achieving a more pronounced effect and delay the emergence of drug resistance. There may also be a commercial value in multi-targeted KIs because one drug can theoretically replace two or three single targeted agents, thereby lowering the cost of treatment.
In recent years, three drugs thought to inhibit multiple kinases entered the market (Sutent, Nexavar and Tykerb), and are seen as a validation of this approach. Interestingly, even kinase inhibitors which were developed as single kinase inhibitors, often have some degree of activity against additional kinases, whether clinically relevant or not. A good example for that might be Gleevec, which had started as a single kinase inhibitor, but then was reborn as an inhibitor of at least two other kinases. At the moment, Exelixis seems to favor the multi-kinase inhibitors approach, with 7 multi-targeted and 4 single-targeted kinase inhibitors in the clinic.
Although there is a wide consensus that simultaneous inhibition of multiple targets may optimize the use of targeted therapies, some question the benefit of multi-targeted inhibitors pointing at several issues. Manipulating several targets at once adds more complexity, on top of the intrinsic complexity of developing a drug against one target. More targets means more variables and uncertainty in terms of the choosing the right combination, the degree of inhibition of each target and the safety profile.
The promiscuous activity of multi-targeted kinase inhibitors often leads to increased toxicity, so these agents are not likely to have an ideal safety profile and consequently may not be given in sufficiently high doses. In addition, it is not always clear whether the clinical activity is due to inhibition of one or multiple targets, and the role each target plays. Most of Nexavar’s activity, for instance, is probably not the result of inhibition of the initial primary target for which the compound was developed, but due to a secondary target. As a result, there are some who believe that simultaneous inhibition of multiple targets should be done with several single-targeted agents rather than with one multi-targeted agent.
Like everything else in drug development, there is no clear solution for this debate, as there is probably more than one way of fighting cancer with kinase inhibitors. The right approach should be chosen on a case by case basis, depending on the specific drug, patient characteristics and the target itself.
Notwithstanding this debate, choosing the right targets is the first, and perhaps the most important step in the process. There is a tradeoff between the extent of clinical validation and the competitive landscape for each target.
Developing drugs that hit validated targets decreases the risk of failure on the one hand, but also involves fierce competition from approved and investigational agents. Examples for two highly validated targets may be EGFR (targeted by Tarceva and Iressa) and Bcr-Abl (targeted by Gleevec, Sprycel and Tasigna). Dethroning an approved drug is a challenging task, not to mention potential competition from compounds in development, so even if a company has a drug that works, it will not necessarily succeed in getting market share.
It is important to understand, though, that presence of approved agents does not necessarily hamper the potential of new compounds. Acknowledging that advanced cancer is almost impossible to cure, the industry is now talking about making it a chronic disease by using many lines of therapy. Kinase inhibitors’ inability to cure the disease and the emergence of resistance creates the need for developing second and third lines of therapy as well as combination treatments. There is, for instance, room for EGFR inhibitors that are active in Tarceva resistant patients or can be given in combination with Tarceva to improve activity and delay the onset of resistance.
Targets for which there are no marketed drugs involve more risk, but also represent the potential of opening up new markets with little or no competition. Exelixis decided to focus on kinases that are still not clinically validated, but such that have a clear role in cancer biology, backed up by a large body of scientific evidence. These targets include MET, PI3K, Hedgehog and MEK. Obviously, there are plenty of other compounds in development that target these kinases, and it is still premature to assess how Exelixis will fare with the competition, but thanks to its size and focus, it managed to be at the forefront of these exciting field.
Exelixis’s approach of targeting the right patient populations and the fact that some of its compounds inhibit multiple kinases may help it differentiate its compounds. In some cases, Exelixis further decreases the risk by developing two distinct compounds against the same target. For example, XL147 and XL765 both inhibit PI3K but the latter inhibits an additional kinase in the same signaling pathway - mTOR. On top of being an independent attempt of striking PI3K, each compound represents a different approach for inhibiting the same pathway in terms of the ideal number of targets to hit in this pathway. It is still unclear which approach is superior, and in which cases, so pursuing both routes in parallel sounds like a good idea.
Summary
Exelixis operates in one of the most promising yet competitive markets in the pharmaceutical industry, where there are always multiple candidates against any given target. The company is not nor will it ever be the undisputed leader of the KI market; however, it managed to distinguish itself as a hybrid between a small biotech and a large pharmaceutical company.
On the one hand, it is an extremely focused company with one of the leading R&D teams in the industry. On the other, it has a broad and diverse pipeline of promising compounds that may be applicable to a wide array of cancers. Most of these compounds will fail either due to poor performance or competing agents, but overall, Exelixis should have enough shots on goal in order to make it.
Disclosure: Author is long EXEL, ISIS, IMGN & SGEN.
***********
The cash issue is the most important factor in thenear term, no doubt. I will try to touch on that matter in the future, but that's an issue everybody including company's mangement is aware of. I expect they'll start to monetizing their portfolio in Q4, after the GSK decision(s). I hear they are very close to siging a deal for one of their PI3K compounds, which are not part the GSK collaboration.
mfg ipollit[/b]
Antwort auf Beitrag Nr.: 35.029.560 von ipollit am 08.09.08 21:58:10die aktuelle Pipeline von EXEL und ihre Targets...
von den mit (*) kann GSK noch ein Programm für die eigene Entwicklung auswählen.


mfg ipollit
von den mit (*) kann GSK noch ein Programm für die eigene Entwicklung auswählen.
mfg ipollit
Antwort auf Beitrag Nr.: 35.029.628 von ipollit am 08.09.08 22:05:15zum Vergleich die aktuellen unverpartnerten Programme und Targets von Array Biopharma (ARRY), die ähnlich wie EXEL vorgehen...

Krebs-Targets...

Entzündungs-Targets...

mfg ipollit
Krebs-Targets...
Entzündungs-Targets...
mfg ipollit
NicOx zweite PIII zu Naproxcinod mit positivem Ergebnis...
NicOx announces second naproxcinod pivotal phase 3 study (302) meets efficacy primary endpoints and supports non-detrimental blood pressure effect
Last update: 1:43 a.m. EDT Sept. 15, 2008
SOPHIA ANTIPOLIS, FRANCE, Sep 15, 2008 (MARKET WIRE via COMTEX) -- September 15, 2008. Sophia Antipolis, France. www.nicox.com
NicOx S.A. (Euronext Paris: COX) today announced successful top-line results from the second phase 3 study for naproxcinod in 1020 patients with osteoarthritis of the knee (the 302 study). Both doses of naproxcinod (750 mg and 375 mg bid) met the three co-primary efficacy endpoints at week-13 (p < 0.001). The study also comfortably met the main secondary endpoint, demonstrating that naproxcinod 750 mg bid was statistically non-inferior to naproxen 500 mg bid on the WOMAC(TM) pain and function subscales at week-13 and 26. Naproxcinod is the first compound in the new Cyclooxygenase-Inhibiting Nitric Oxide-Donator (CINOD) class of anti-inflammatory agents, which NicOx is developing as a drug for the treatment of the signs and symptoms of osteoarthritis.
NicOx' phase 3 clinical program for naproxcinod consists of three pivotal trials (including the previously completed 301 study in osteoarthritis of the knee and the ongoing 303 study in osteoarthritis of the hip, in addition to the 302 study). Overall, the results of the 302 study support naproxcinod's non-detrimental effect on blood pressure and are consistent with those observed in the 301 study, with naproxcinod 750 mg bid showing a numerical reduction in systolic and diastolic blood pressure at week-13 and 26, compared to baseline and naproxen 500 mg bid. Existing non-steroidal anti-inflammatory agents (NSAIDs) such as ibuprofen and naproxen have the tendency to raise blood pressure, which is a side effect of particular concern in the osteoarthritis population.
A post hoc pooled analysis of the blood pressure data from the 301 and 302 studies showed a statistically significant reduction for naproxcinod 750 mg bid, compared to naproxen 500 mg bid, in terms of the mean change from baseline at week-13, of 2.3 mmHg (p < > =0.004) for systolic blood pressure and 1.1 mmHg (p < > =0.034) for diastolic blood pressure. In the planning of the phase 3 program, NicOx had foreseen the pooling of office blood pressure measurements (OBPMs) from more than one study in order to obtain the necessary statistical power to correctly assess the effects of naproxcinod on blood pressure, compared to naproxen. A pre-specified analysis of the pooled blood pressure data from the three phase 3 studies will be conducted following the completion of the third phase 3 trial (the 303 study) in the fourth quarter of 2008.
Michele Garufi, Chairman and CEO of NicOx, commented: "We are impressed by the strong efficacy results observed in this study, which have replicated the positive results of the 301 trial and clearly demonstrated non-inferiority to naproxen at 26 weeks. The results of these two pivotal studies give us increasing confidence that naproxcinod will meet regulatory requirements for approval in the United States and Europe. Moreover, based on the results of the pooled analysis on blood pressure from these studies, we firmly believe that naproxcinod has the potential to fulfill the current medical need for an efficacious anti-inflammatory agent with no detrimental impact on blood pressure."
Positive efficacy results in comparison to placebo and naproxen
As in the 301 study, both naproxcinod doses (375 mg bid and 750 mg bid) were shown to be superior to placebo on the three co-primary efficacy endpoints of the 302 trial, the WOMAC(TM) pain subscale, the WOMAC(TM) function subscale and patients' overall rating of disease status, with these being highly statistically significant (p < 0.001) in terms of the mean change from baseline at week-13. Superiority comparisons versus placebo on these three co-primary endpoints in two well designed, well controlled trials conform to U.S. Food and Drug Administration (FDA) guidance for demonstrating the efficacy of new drugs for the treatment of the signs and symptoms of osteoarthritis.
The main secondary endpoint of the trial assessed the efficacy of naproxcinod 750 mg bid to naproxen 500 mg bid at week-13 and 26 in terms of the mean change from baseline in the WOMAC(TM) pain and function subscales. Naproxcinod 750 mg bid comfortably met the secondary endpoint of being statistically non-inferior to naproxen 500 mg bid on these two parameters. Although not a pre-specified secondary endpoint of the study, a post hoc analysis showed that naproxcinod 375 mg bid was also statistically non-inferior to naproxen 500 mg bid at week-26 on these two parameters. Non-inferiority comparisons to existing products are required by the European Medicines Agency (EMEA) for demonstrating the efficacy of new drugs.
Results support favorable blood pressure profile versus naproxen
During the trial, patients' blood pressure was measured using office blood pressure measurements (OBPM) at each visit to the clinical center, using a standardized, well controlled approach. A pre-specified analysis compared naproxcinod to naproxen, in terms of the mean change from baseline in systolic and diastolic blood pressure at baseline and weeks-2, 6, 13, 15 and 26. The results support naproxcinod's non-detrimental effect on blood pressure and are consistent with those observed in the 301 study with both doses of naproxcinod showing a numerical reduction in systolic and diastolic blood pressure, compared to baseline and naproxen 500 mg bid, which was sustained over the vast majority of the time points.
No safety concerns up to week-26
There were no safety concerns relating to naproxcinod treatment up to week-26 and all the active treatment arms were similar to placebo in terms of the percentage of patients with one or more adverse event at week-13. At week-26, the percentage of patients with one or more adverse event was similar for each of the study drugs. The number of serious adverse events was low and evenly distributed among the treatment groups.
NOTE: NicOx entered into a full-service agreement for the conduct of the 302 study with Premier Research Group Limited, a global CRO specializing in the conduct of clinical trials in pain indications and other important therapeutic areas. The study is a 53-week, randomized, double-blind, efficacy and safety trial in which 1020 patients with osteoarthritis of the knee were enrolled at around 150 clinical sites throughout the United States. Patients were randomized to one of the following treatment groups: naproxcinod 375 mg bid (52 weeks), naproxcinod 750 mg bid (52 weeks), naproxen 500 mg bid (52 weeks) and placebo bid during the first 13 weeks. After week-13, the placebo treated patients were randomized in a blinded fashion to either naproxcinod 375 mg bid or naproxcinod 750 mg bid for the remainder of the trial (39 weeks). Patients had primary osteoarthritis of the knee of at least 3 months duration. The general safety and tolerability of naproxcinod are still being assessed until week-52 and the trial has a 1 week post-treatment safety period. For further details of the 302 study design, see the NicOx press release of April 3, 2007.
mfg ipollit
NicOx announces second naproxcinod pivotal phase 3 study (302) meets efficacy primary endpoints and supports non-detrimental blood pressure effect
Last update: 1:43 a.m. EDT Sept. 15, 2008
SOPHIA ANTIPOLIS, FRANCE, Sep 15, 2008 (MARKET WIRE via COMTEX) -- September 15, 2008. Sophia Antipolis, France. www.nicox.com
NicOx S.A. (Euronext Paris: COX) today announced successful top-line results from the second phase 3 study for naproxcinod in 1020 patients with osteoarthritis of the knee (the 302 study). Both doses of naproxcinod (750 mg and 375 mg bid) met the three co-primary efficacy endpoints at week-13 (p < 0.001). The study also comfortably met the main secondary endpoint, demonstrating that naproxcinod 750 mg bid was statistically non-inferior to naproxen 500 mg bid on the WOMAC(TM) pain and function subscales at week-13 and 26. Naproxcinod is the first compound in the new Cyclooxygenase-Inhibiting Nitric Oxide-Donator (CINOD) class of anti-inflammatory agents, which NicOx is developing as a drug for the treatment of the signs and symptoms of osteoarthritis.
NicOx' phase 3 clinical program for naproxcinod consists of three pivotal trials (including the previously completed 301 study in osteoarthritis of the knee and the ongoing 303 study in osteoarthritis of the hip, in addition to the 302 study). Overall, the results of the 302 study support naproxcinod's non-detrimental effect on blood pressure and are consistent with those observed in the 301 study, with naproxcinod 750 mg bid showing a numerical reduction in systolic and diastolic blood pressure at week-13 and 26, compared to baseline and naproxen 500 mg bid. Existing non-steroidal anti-inflammatory agents (NSAIDs) such as ibuprofen and naproxen have the tendency to raise blood pressure, which is a side effect of particular concern in the osteoarthritis population.
A post hoc pooled analysis of the blood pressure data from the 301 and 302 studies showed a statistically significant reduction for naproxcinod 750 mg bid, compared to naproxen 500 mg bid, in terms of the mean change from baseline at week-13, of 2.3 mmHg (p < > =0.004) for systolic blood pressure and 1.1 mmHg (p < > =0.034) for diastolic blood pressure. In the planning of the phase 3 program, NicOx had foreseen the pooling of office blood pressure measurements (OBPMs) from more than one study in order to obtain the necessary statistical power to correctly assess the effects of naproxcinod on blood pressure, compared to naproxen. A pre-specified analysis of the pooled blood pressure data from the three phase 3 studies will be conducted following the completion of the third phase 3 trial (the 303 study) in the fourth quarter of 2008.
Michele Garufi, Chairman and CEO of NicOx, commented: "We are impressed by the strong efficacy results observed in this study, which have replicated the positive results of the 301 trial and clearly demonstrated non-inferiority to naproxen at 26 weeks. The results of these two pivotal studies give us increasing confidence that naproxcinod will meet regulatory requirements for approval in the United States and Europe. Moreover, based on the results of the pooled analysis on blood pressure from these studies, we firmly believe that naproxcinod has the potential to fulfill the current medical need for an efficacious anti-inflammatory agent with no detrimental impact on blood pressure."
Positive efficacy results in comparison to placebo and naproxen
As in the 301 study, both naproxcinod doses (375 mg bid and 750 mg bid) were shown to be superior to placebo on the three co-primary efficacy endpoints of the 302 trial, the WOMAC(TM) pain subscale, the WOMAC(TM) function subscale and patients' overall rating of disease status, with these being highly statistically significant (p < 0.001) in terms of the mean change from baseline at week-13. Superiority comparisons versus placebo on these three co-primary endpoints in two well designed, well controlled trials conform to U.S. Food and Drug Administration (FDA) guidance for demonstrating the efficacy of new drugs for the treatment of the signs and symptoms of osteoarthritis.
The main secondary endpoint of the trial assessed the efficacy of naproxcinod 750 mg bid to naproxen 500 mg bid at week-13 and 26 in terms of the mean change from baseline in the WOMAC(TM) pain and function subscales. Naproxcinod 750 mg bid comfortably met the secondary endpoint of being statistically non-inferior to naproxen 500 mg bid on these two parameters. Although not a pre-specified secondary endpoint of the study, a post hoc analysis showed that naproxcinod 375 mg bid was also statistically non-inferior to naproxen 500 mg bid at week-26 on these two parameters. Non-inferiority comparisons to existing products are required by the European Medicines Agency (EMEA) for demonstrating the efficacy of new drugs.
Results support favorable blood pressure profile versus naproxen
During the trial, patients' blood pressure was measured using office blood pressure measurements (OBPM) at each visit to the clinical center, using a standardized, well controlled approach. A pre-specified analysis compared naproxcinod to naproxen, in terms of the mean change from baseline in systolic and diastolic blood pressure at baseline and weeks-2, 6, 13, 15 and 26. The results support naproxcinod's non-detrimental effect on blood pressure and are consistent with those observed in the 301 study with both doses of naproxcinod showing a numerical reduction in systolic and diastolic blood pressure, compared to baseline and naproxen 500 mg bid, which was sustained over the vast majority of the time points.
No safety concerns up to week-26
There were no safety concerns relating to naproxcinod treatment up to week-26 and all the active treatment arms were similar to placebo in terms of the percentage of patients with one or more adverse event at week-13. At week-26, the percentage of patients with one or more adverse event was similar for each of the study drugs. The number of serious adverse events was low and evenly distributed among the treatment groups.
NOTE: NicOx entered into a full-service agreement for the conduct of the 302 study with Premier Research Group Limited, a global CRO specializing in the conduct of clinical trials in pain indications and other important therapeutic areas. The study is a 53-week, randomized, double-blind, efficacy and safety trial in which 1020 patients with osteoarthritis of the knee were enrolled at around 150 clinical sites throughout the United States. Patients were randomized to one of the following treatment groups: naproxcinod 375 mg bid (52 weeks), naproxcinod 750 mg bid (52 weeks), naproxen 500 mg bid (52 weeks) and placebo bid during the first 13 weeks. After week-13, the placebo treated patients were randomized in a blinded fashion to either naproxcinod 375 mg bid or naproxcinod 750 mg bid for the remainder of the trial (39 weeks). Patients had primary osteoarthritis of the knee of at least 3 months duration. The general safety and tolerability of naproxcinod are still being assessed until week-52 and the trial has a 1 week post-treatment safety period. For further details of the 302 study design, see the NicOx press release of April 3, 2007.
mfg ipollit
http://www.stockhouse.com/Columnists/2008/September/15/Expir…
Expiring patents will ignite biotech boom
9/15/2008 9:12:34 AM | Andrew Mickey
A buyout frenzy could spark a very bull market in biotech.
It’s tough to find anything positive to talk about in this market.
The U.S. government stepped in and assigned Fannie Mae (NYSE: FNM, Stock Forum)and Freddie Mac (NYSE: FRE, Stock Forum) a value of zero through governmental decree.
Even a cutback in oil production from OPEC can’t stop oil prices from falling. Gold, silver, and fertilizer stocks are falling too. Even Brazil’s stock market, the top-performing BRIC nation of 2008, is starting to fall.
It’s bad out there for most industries. But there’s no sector worse off than the pharmaceutical sector. Big Pharma stocks like Pfizer (NYSE: PFE, Stock Forum), Merck (NYSE: MRK, Stock Forum), and GlaxoSmithKline (NYSE: GSK, Stock Forum) are having a rough time and it looks like more stormy weather ahead. And these companies are giants headed for a fall.
But here’s the thing about the drug industry, bad times for the big guys can mean good times for the little guys. And that means great profits for savvy investors. Right now, Big Pharma is searching for new revenue streams, and that is good news for the shareholders of smaller, innovative drug makers.
CNN summed Big Pharma’s problems up best when it said, “[The industry is] like rearranging chairs on the Titanic.”
Frankly, the situation is bad… real bad. Even nine-digit lobbying budgets and their hundreds of Ivy League-educated lawyers aren’t going to be able to stop it.
The cause of the problem is complacency. You see, Big Pharma lives on “blockbuster” drugs. A blockbuster drug is simply any drug that generates at least $1 billion in sales each year.
And it’s those drugs that make the big profits for the multi-billion-dollar pharma companies like Pfizer, Merck, and GlaxoSmithKline.
On average, it takes about $800 million to develop a drug. That takes a moment to digest. It’s an astronomical price tag for a product that is difficult to “market test.” The $800 million covers everything from early pre-clinical trials, the three phases of FDA approvals, and marketing costs. As a result, it takes a big success to make any money in the drug industry.
So for these big drug companies, $1 billion is basically the break even point.
Keep in mind, that’s not including the costs to manufacture each pill… and the costs of any lawsuits, production delays, competition, patent disputes, and everything else that seem to pile up.
There’s another significant challenge the Big Pharma companies have to deal with: the length of the patent. New drugs are only patented for 20 years. That sounds like a generous amount of time. If they had 20 years, a drug company would have to sell only about $40 million of a drug to recoup those costs of developing a new one. But investors are not typically patient enough to operate on that time-line.
At the 20-year point, the patent expires and anyone can make the drug and sell it. The generic drug industry is founded on manufacturing drugs after the patent runs out. Generic drug makers wait for the patents to expire, take the formula that costs hundreds of billions of dollars to develop, and reproduce.
Since they have none of the $800 million in research costs to recover, they can easily undercut the prices of the original developer of the drug. The generics can cost as much as 80% to 90% less than brand name drugs. Consumers inevitably turn to the cheaper version.
But we’re discounting one technicality. According to U.S. patent law, patented drugs are protected from competition for 20 years. In reality, drug companies don’t have nearly that long.
The clock starts ticking when the patent is filed. That’s before the safety and development process even begins.
Since that process usually takes at least a decade, drug companies don’t have anything close to 20 years… they usually have only seven to 10 years to sell their drugs at top prices. They’re protected from competition by patents for a very short time. And once the patents expire, the generic manufacturers move in and take sales away.
In the next few years, the worst possible thing is going to happen to Big Pharma. The industry that needs vast amounts of cash to fund high-risk projects knowing any payoff is 10 years away (at the earliest) is going to get cut off from their lifeblood….
The number of drugs going “off patent” in the next few years is alarming. According to IMH Health, more than $60 billion worth of drugs are going off patent by 2011.
Take Fosamax for example. Fosamax is Merck’s current top-selling drug. It’s designed to help treat osteoporosis by making bones stronger. It brings in more than $3 billion in sales to the company each year. It was a huge success for Merck….
Emphasis on was. The Fosamax patent expired in February of 2008.
Fosamax accounted for more than 12% of one of the largest pharmaceutical companies in the world! Now, it’s all gone. Anybody can take the secret formula that made Fosamax a success, directly copy it, and resell it for as cheap as possible.
Fosamax is just one example of the huge impact of a drug going off patent. It’s happening all across Big Pharma.
GlaxoSmithKline’s Advair came off patent in February 2008. Advair accounted for 15% of annual sales for GSK.
Top selling anti-depressant Effexor, which accounts for $3.7 billion in annual sales for Wyeth (NYSE: WYE, Stock Forum), has gone off patent too. Last June, when the patent expired, 6% of Wyeth’s business was eliminated.
Johnson & Johnson’s (NYSE: JNJ, Stock Forum) Topamax, which racks up $2.5 billion in annual sales, is going off patent in September. AstraZeneca’s (NYSE: AZN, Stock Forum) $1.2 billion loses patent protection in October….
You get the picture. It’s happening now, and more and more drugs are going to go off patent in the next two years.
Big Pharma is going to be forced to buy small biotech companies just to survive and the buyout frenzy could spark a very bull market in biotech.
In fact, it has already started. Early last week, a rumor of an “imminent” takeover by Pfizer of Bayer sent shares of the German drug maker climbing 5%. Forbes reports the top cause of the buyout is Bayer’s extensive pipeline of new drugs in development.
Although any Pfizer/Bayer combo is unlikely and far from a perfect fit, the excitement surrounding it proves how desperate Big Pharma is becoming.
At the end of the day, the investing principals we stick to at the Prosperity Dispatch are pretty simple. You buy something (a stock) at a low price and hope to sell it at a higher price. And right now, you can buy small biotech stocks at a low price that are good candidates to be bought out by deep-pocketed buyers at a higher price.
Good investing,
Andrew Mickey
mfg ipollit
Expiring patents will ignite biotech boom
9/15/2008 9:12:34 AM | Andrew Mickey
A buyout frenzy could spark a very bull market in biotech.
It’s tough to find anything positive to talk about in this market.
The U.S. government stepped in and assigned Fannie Mae (NYSE: FNM, Stock Forum)and Freddie Mac (NYSE: FRE, Stock Forum) a value of zero through governmental decree.
Even a cutback in oil production from OPEC can’t stop oil prices from falling. Gold, silver, and fertilizer stocks are falling too. Even Brazil’s stock market, the top-performing BRIC nation of 2008, is starting to fall.
It’s bad out there for most industries. But there’s no sector worse off than the pharmaceutical sector. Big Pharma stocks like Pfizer (NYSE: PFE, Stock Forum), Merck (NYSE: MRK, Stock Forum), and GlaxoSmithKline (NYSE: GSK, Stock Forum) are having a rough time and it looks like more stormy weather ahead. And these companies are giants headed for a fall.
But here’s the thing about the drug industry, bad times for the big guys can mean good times for the little guys. And that means great profits for savvy investors. Right now, Big Pharma is searching for new revenue streams, and that is good news for the shareholders of smaller, innovative drug makers.
CNN summed Big Pharma’s problems up best when it said, “[The industry is] like rearranging chairs on the Titanic.”
Frankly, the situation is bad… real bad. Even nine-digit lobbying budgets and their hundreds of Ivy League-educated lawyers aren’t going to be able to stop it.
The cause of the problem is complacency. You see, Big Pharma lives on “blockbuster” drugs. A blockbuster drug is simply any drug that generates at least $1 billion in sales each year.
And it’s those drugs that make the big profits for the multi-billion-dollar pharma companies like Pfizer, Merck, and GlaxoSmithKline.
On average, it takes about $800 million to develop a drug. That takes a moment to digest. It’s an astronomical price tag for a product that is difficult to “market test.” The $800 million covers everything from early pre-clinical trials, the three phases of FDA approvals, and marketing costs. As a result, it takes a big success to make any money in the drug industry.
So for these big drug companies, $1 billion is basically the break even point.
Keep in mind, that’s not including the costs to manufacture each pill… and the costs of any lawsuits, production delays, competition, patent disputes, and everything else that seem to pile up.
There’s another significant challenge the Big Pharma companies have to deal with: the length of the patent. New drugs are only patented for 20 years. That sounds like a generous amount of time. If they had 20 years, a drug company would have to sell only about $40 million of a drug to recoup those costs of developing a new one. But investors are not typically patient enough to operate on that time-line.
At the 20-year point, the patent expires and anyone can make the drug and sell it. The generic drug industry is founded on manufacturing drugs after the patent runs out. Generic drug makers wait for the patents to expire, take the formula that costs hundreds of billions of dollars to develop, and reproduce.
Since they have none of the $800 million in research costs to recover, they can easily undercut the prices of the original developer of the drug. The generics can cost as much as 80% to 90% less than brand name drugs. Consumers inevitably turn to the cheaper version.
But we’re discounting one technicality. According to U.S. patent law, patented drugs are protected from competition for 20 years. In reality, drug companies don’t have nearly that long.
The clock starts ticking when the patent is filed. That’s before the safety and development process even begins.
Since that process usually takes at least a decade, drug companies don’t have anything close to 20 years… they usually have only seven to 10 years to sell their drugs at top prices. They’re protected from competition by patents for a very short time. And once the patents expire, the generic manufacturers move in and take sales away.
In the next few years, the worst possible thing is going to happen to Big Pharma. The industry that needs vast amounts of cash to fund high-risk projects knowing any payoff is 10 years away (at the earliest) is going to get cut off from their lifeblood….
The number of drugs going “off patent” in the next few years is alarming. According to IMH Health, more than $60 billion worth of drugs are going off patent by 2011.
Take Fosamax for example. Fosamax is Merck’s current top-selling drug. It’s designed to help treat osteoporosis by making bones stronger. It brings in more than $3 billion in sales to the company each year. It was a huge success for Merck….
Emphasis on was. The Fosamax patent expired in February of 2008.
Fosamax accounted for more than 12% of one of the largest pharmaceutical companies in the world! Now, it’s all gone. Anybody can take the secret formula that made Fosamax a success, directly copy it, and resell it for as cheap as possible.
Fosamax is just one example of the huge impact of a drug going off patent. It’s happening all across Big Pharma.
GlaxoSmithKline’s Advair came off patent in February 2008. Advair accounted for 15% of annual sales for GSK.
Top selling anti-depressant Effexor, which accounts for $3.7 billion in annual sales for Wyeth (NYSE: WYE, Stock Forum), has gone off patent too. Last June, when the patent expired, 6% of Wyeth’s business was eliminated.
Johnson & Johnson’s (NYSE: JNJ, Stock Forum) Topamax, which racks up $2.5 billion in annual sales, is going off patent in September. AstraZeneca’s (NYSE: AZN, Stock Forum) $1.2 billion loses patent protection in October….
You get the picture. It’s happening now, and more and more drugs are going to go off patent in the next two years.
Big Pharma is going to be forced to buy small biotech companies just to survive and the buyout frenzy could spark a very bull market in biotech.
In fact, it has already started. Early last week, a rumor of an “imminent” takeover by Pfizer of Bayer sent shares of the German drug maker climbing 5%. Forbes reports the top cause of the buyout is Bayer’s extensive pipeline of new drugs in development.
Although any Pfizer/Bayer combo is unlikely and far from a perfect fit, the excitement surrounding it proves how desperate Big Pharma is becoming.
At the end of the day, the investing principals we stick to at the Prosperity Dispatch are pretty simple. You buy something (a stock) at a low price and hope to sell it at a higher price. And right now, you can buy small biotech stocks at a low price that are good candidates to be bought out by deep-pocketed buyers at a higher price.
Good investing,
Andrew Mickey
mfg ipollit
http://www.ft.com/cms/s/0/9d620a56-85d6-11dd-a1ac-0000779fd1…
Drugmakers sweeten deals for biotech groups
By Andrew Jack
Published: September 19 2008 04:27 | Last updated: September 19 2008 04:27
The global credit crunch and the equity meltdown may have caused pain to companies in most sectors, but it has had unexpected benefits for cash-rich established drugmakers such as Shire, the Ireland-based speciality pharmaceutical business.
“It has strengthened our negotiating position,” says Angus Russell, chief executive, who has completed a string of deals in recent years, including a friendly takeover currently being completed of Jerini, a Berlin-based biotech company.
Jerini’s relatively low stock market value made outright purchase cheaper and simpler than arranging complex licensing terms for its peptide-based medicines that Shire sought. In other cases, fewer options for funding have made biotech companies keener to do deals quickly with it, where they previously had held out for better pricing.
The pattern has been repeated elsewhere and has helped shift the balance of power away from biotech companies in their discussions with larger pharmaceutical groups that are willing to pay high prices as they compete in their search to find future drugs to replenish thin portfolios.
“In the past few years, it was a sellers’ market driving up valuations for less and less mature technology,” says Tibur Papp, head of advisory at PharmaVentures, a UK-based consultancy. “Now the environment is changing and with a significant impact on biotech finance, it has become more difficult to find partners and good deals.”
The tough background, reinforced by the collapse of companies such as Ardana and Phoqus, has sparked reflection on the need for further industry consolidation, such as Thursday’s merger of BTG and Protherics, a move that advisers to both companies stress was conducted from a position of strength.
Better solutions have been found by smaller companies that have given up their hopes to remain independent: Acambis, the vaccines developer, is being acquired by Sanofi-Aventis of France, and Novartis of Switzerland recently acquired Speedel .
More broadly, Neil Mackison, head of European healthcare investment banking at Piper Jaffrey, a middle market investment bank, says: “Companies that are running out of money are looking for new strategies.”
With scant prospect for initial public offerings or follow-up equity financing, and venture capitalists seeking returns of 25-30 per cent, he says that some have turned to “venture debt”, offering coupons that he says are typically “in the high teens”.
Those lucky enough to have medicines on the market are doing more deals with specialist royalty companies to provide cash up-front in exchange for the rights to future sales.
That leaves two options: licensing earlier with partners, at a point when less clear-cut clinical trial data mean the prospect of lower prices; and takeovers. “Mergers and acquisitions are definitely top of the agenda, far more than in the past four or five years,” he says.
Given the increasing recognition by large pharmaceutical groups of the need to seek products outside their own laboratories, and sector-wide concerns about a series of existing best-selling products coming off patent in the next few years, biotech companies with promising products are still able to command good prices.
“Products remain the lifeblood of the industry, and the opportunities for low- and mid-hanging fruit are getting fewer,” stresses Robin Campbell, an analyst with Jefferies. “There is a degree of corporate anorexia within big pharma, with their research centres being picked apart, and that means in-licensing and business development people have greater say.”
He also points out that relative valuations of the more promising biotech and pharmaceutical companies have been improving recently, as investors divest from financial institutions and seek more defensive stocks.
Aisling Burnand, head of the BioIndustry Association, the UK trade body, argues: “The fundamentals of the sector are still quite strong. Biotechs have not necessarily been worse impacted than any other sector and are more seasoned in belt-tightening than many.”
But many biotech businesses have few products of uncertain quality. John Mayo, head of Celtic Pharma, a company that buys experimental medicines for resale, argues that big pharma remains suspicious because such drugs are often “hairy”, supported by insufficient clinical trial data to win regulatory approval.
As an adviser to the sector says: “The biotech sector needs to consolidate. There are too many bit players with a single product in the clinic, all the infrastructure costs of PLCs, and a £400,000 ($722,597) salary for the chief executive. But egos and valuations get in the way.”
mfg ipollit
Drugmakers sweeten deals for biotech groups
By Andrew Jack
Published: September 19 2008 04:27 | Last updated: September 19 2008 04:27
The global credit crunch and the equity meltdown may have caused pain to companies in most sectors, but it has had unexpected benefits for cash-rich established drugmakers such as Shire, the Ireland-based speciality pharmaceutical business.
“It has strengthened our negotiating position,” says Angus Russell, chief executive, who has completed a string of deals in recent years, including a friendly takeover currently being completed of Jerini, a Berlin-based biotech company.
Jerini’s relatively low stock market value made outright purchase cheaper and simpler than arranging complex licensing terms for its peptide-based medicines that Shire sought. In other cases, fewer options for funding have made biotech companies keener to do deals quickly with it, where they previously had held out for better pricing.
The pattern has been repeated elsewhere and has helped shift the balance of power away from biotech companies in their discussions with larger pharmaceutical groups that are willing to pay high prices as they compete in their search to find future drugs to replenish thin portfolios.
“In the past few years, it was a sellers’ market driving up valuations for less and less mature technology,” says Tibur Papp, head of advisory at PharmaVentures, a UK-based consultancy. “Now the environment is changing and with a significant impact on biotech finance, it has become more difficult to find partners and good deals.”
The tough background, reinforced by the collapse of companies such as Ardana and Phoqus, has sparked reflection on the need for further industry consolidation, such as Thursday’s merger of BTG and Protherics, a move that advisers to both companies stress was conducted from a position of strength.
Better solutions have been found by smaller companies that have given up their hopes to remain independent: Acambis, the vaccines developer, is being acquired by Sanofi-Aventis of France, and Novartis of Switzerland recently acquired Speedel .
More broadly, Neil Mackison, head of European healthcare investment banking at Piper Jaffrey, a middle market investment bank, says: “Companies that are running out of money are looking for new strategies.”
With scant prospect for initial public offerings or follow-up equity financing, and venture capitalists seeking returns of 25-30 per cent, he says that some have turned to “venture debt”, offering coupons that he says are typically “in the high teens”.
Those lucky enough to have medicines on the market are doing more deals with specialist royalty companies to provide cash up-front in exchange for the rights to future sales.
That leaves two options: licensing earlier with partners, at a point when less clear-cut clinical trial data mean the prospect of lower prices; and takeovers. “Mergers and acquisitions are definitely top of the agenda, far more than in the past four or five years,” he says.
Given the increasing recognition by large pharmaceutical groups of the need to seek products outside their own laboratories, and sector-wide concerns about a series of existing best-selling products coming off patent in the next few years, biotech companies with promising products are still able to command good prices.
“Products remain the lifeblood of the industry, and the opportunities for low- and mid-hanging fruit are getting fewer,” stresses Robin Campbell, an analyst with Jefferies. “There is a degree of corporate anorexia within big pharma, with their research centres being picked apart, and that means in-licensing and business development people have greater say.”
He also points out that relative valuations of the more promising biotech and pharmaceutical companies have been improving recently, as investors divest from financial institutions and seek more defensive stocks.
Aisling Burnand, head of the BioIndustry Association, the UK trade body, argues: “The fundamentals of the sector are still quite strong. Biotechs have not necessarily been worse impacted than any other sector and are more seasoned in belt-tightening than many.”
But many biotech businesses have few products of uncertain quality. John Mayo, head of Celtic Pharma, a company that buys experimental medicines for resale, argues that big pharma remains suspicious because such drugs are often “hairy”, supported by insufficient clinical trial data to win regulatory approval.
As an adviser to the sector says: “The biotech sector needs to consolidate. There are too many bit players with a single product in the clinic, all the infrastructure costs of PLCs, and a £400,000 ($722,597) salary for the chief executive. But egos and valuations get in the way.”
mfg ipollit
RIGL(R788, PIIb, SYK), INCY(INCB018424, PII, JAK1/2), EXEL(XL019, PI, JAK2)... orale Mittel gegen RA:
CS: Rigel Pharmaceuticals Inc. (RIGL) Outperform [V] M. Aberman
CP: US$ 24.90 TP: US$ 37 CAP: US$ 909.1m
ACR 2008 - Landscape for Oral RA Agents
• Conclusion: There is not much new to the ACR abstract for R788, which contains the 12-week data for R788 in RA. That said, R788's efficacy data continues to compare very favorably with the other oral kinase inhibitors targeting RA (e.g. Pfizer's JAK-3 and Incyte's JAK 1/2 inhibitor) and there were no new safety signals reported. Pfizer's lack of dose response could limit efficacy to what we have already seen, but could allow a lower dose to be used with potentially less toxicity. Incyte's data look encouraging; however, we believe the data are too early (4 weeks) and too sparse (12 patients) to draw any firm conclusions. We think all of these agents/targets show promise and given the size of the market, there will be room for many players. One potential advantage for R788 is that, to our knowledge, they are the only company that is targeting SYK. On the safety side, both Rigel's R788 and Pfizer's JAK3 have had tolerability signals and we look forward to the plenary session and company-sponsored update discussing one-year safety data of the Phase IIA R788 trial in RA to gain better insight into both efficacy and tolerability.
• What's New? Abstracts from ACR (American College of Rheumatology) 2008 have been released, with new efficacy and safety data kinase inhibitors including Rigel's oral syk kinase inhibitor R788, Pfizer's JAK-3 inhibitor CP-690,550, and Incyte's JAK-1&2 inhibitor INCB018424.
• Implication: We remain Outperform rated with our existing price target of $37. We continue to believe that Rigel is undervalued given its proof of concept efficacy in RA, ITP, and non-Hodgkin's lymphoma. The Phase II data set of R788 in RA, including a full year of safety data, should be another positive for the stock when it is presented at ACR in October, and we view a strong partnership as another potential positive catalyst in late 2008/early 2009
**************
Friedman’s remarks about competitive oral compounds for MF and/or RA from last week’s presentations( I have no idea why he talks about other companies; nothing good can come of it):
(i) exel - - has off-target toxicity issues; now at low doses to avoid that issue; we think that the dose is too low to be efficacious
(ii) targegen (private san diego) - - dosing compound up by factor of 10; very selective for JAK2, not JAK1 or JAK3; JAK1 selectivity is important; compound could make it
(iii) s*bio (Singapore quasi-govt.) - - don’t know much about it
(iv) cytopia ( private Australia) - - preclinical
(v) azn - - preclinical
(vi) pfe - - expect good results; compelling data; good efficacy; key will be therapeutic index and safety profile; oral agents should be as good as biologics; might see immunosuppression because of JAK3; they may try crohn’s as well
(vii) rigl - - pretty good chance of success; narrow therapeutic index; not that selective; clinical efficacy; DMARD qualities; ahead of us; at safe 50 mg, less activity than ours; reasonably good compound; ours and pfe’s will likely be better[/i]
mfg ipollit
CS: Rigel Pharmaceuticals Inc. (RIGL) Outperform [V] M. Aberman
CP: US$ 24.90 TP: US$ 37 CAP: US$ 909.1m
ACR 2008 - Landscape for Oral RA Agents
• Conclusion: There is not much new to the ACR abstract for R788, which contains the 12-week data for R788 in RA. That said, R788's efficacy data continues to compare very favorably with the other oral kinase inhibitors targeting RA (e.g. Pfizer's JAK-3 and Incyte's JAK 1/2 inhibitor) and there were no new safety signals reported. Pfizer's lack of dose response could limit efficacy to what we have already seen, but could allow a lower dose to be used with potentially less toxicity. Incyte's data look encouraging; however, we believe the data are too early (4 weeks) and too sparse (12 patients) to draw any firm conclusions. We think all of these agents/targets show promise and given the size of the market, there will be room for many players. One potential advantage for R788 is that, to our knowledge, they are the only company that is targeting SYK. On the safety side, both Rigel's R788 and Pfizer's JAK3 have had tolerability signals and we look forward to the plenary session and company-sponsored update discussing one-year safety data of the Phase IIA R788 trial in RA to gain better insight into both efficacy and tolerability.
• What's New? Abstracts from ACR (American College of Rheumatology) 2008 have been released, with new efficacy and safety data kinase inhibitors including Rigel's oral syk kinase inhibitor R788, Pfizer's JAK-3 inhibitor CP-690,550, and Incyte's JAK-1&2 inhibitor INCB018424.
• Implication: We remain Outperform rated with our existing price target of $37. We continue to believe that Rigel is undervalued given its proof of concept efficacy in RA, ITP, and non-Hodgkin's lymphoma. The Phase II data set of R788 in RA, including a full year of safety data, should be another positive for the stock when it is presented at ACR in October, and we view a strong partnership as another potential positive catalyst in late 2008/early 2009
**************
Friedman’s remarks about competitive oral compounds for MF and/or RA from last week’s presentations( I have no idea why he talks about other companies; nothing good can come of it):
(i) exel - - has off-target toxicity issues; now at low doses to avoid that issue; we think that the dose is too low to be efficacious
(ii) targegen (private san diego) - - dosing compound up by factor of 10; very selective for JAK2, not JAK1 or JAK3; JAK1 selectivity is important; compound could make it
(iii) s*bio (Singapore quasi-govt.) - - don’t know much about it
(iv) cytopia ( private Australia) - - preclinical
(v) azn - - preclinical
(vi) pfe - - expect good results; compelling data; good efficacy; key will be therapeutic index and safety profile; oral agents should be as good as biologics; might see immunosuppression because of JAK3; they may try crohn’s as well
(vii) rigl - - pretty good chance of success; narrow therapeutic index; not that selective; clinical efficacy; DMARD qualities; ahead of us; at safe 50 mg, less activity than ours; reasonably good compound; ours and pfe’s will likely be better[/i]
mfg ipollit
Genmab...
Genmab / Buy / DKK 308 - Solid impressions on Ofatumumab and EGFr from meeting with SVP Medical Affairs
• Very solid data on Ofatumumab in CLL - ASH presentation to be a strong trigger
• Delays to EGFr data confirmed to be a positive sign
• Good EGFr data could trigger next major deal
Looking forward to ASH in December
Handelsbanken Capital Markets hosted a "Science Session" with Genmab's SVP Medical Affairs Dr. Ole Baadsgaard aimed to discuss the Ofatumumab (anti CD20) programme, the phase III data released in chronic lymphocytic leukemia (CLL) as well as expectations for the phase III programme with Zalutumumab (anti EGFr) in head & neck cancer. There is no doubt in our mind that the phase III data with Ofatumumab in CLL are very strong and solid and that Genmab/GSK appear to be in possession of a very potent and effective antibody. Genmab has still not received confirmation that the data is accepted for presentation at ASH December 6-9 but the company expect data to be accepted. Furthermore we can sense on Dr. Baadsgaard that he would look very much forward to have the data presented at ASH due to the quality of data and strength of results. We believe that data will show that Ofatumumab also works in patients previously treated with Roche/Genentech's blockbuster antibody Rituxan and furthermore that there's a likelihood data could should that Ofatumumab has strong effect even in difficult genotypes with bad disease prognosis. We note that the data, according to Dr. Baadsgaard, surprised all participating parties in the Ofatumumab phase III study including the company as well as haematologists that had hoped for response rates of 30-35% and not the 44-51% as announced.
Looking forward to EGFr data
As argued above we look forward to ASH in December for the Ofatumumab phase III CLL data presentation. We look equally as much forward to the presentation of interim data/headline results for Genmab's EGFr antibody Zalutumumab in head & neck cancer. Dr. Baadsgaard confirmed at our meeting that the numerous delays to release of headline data from the phase III programme should be seen as positive rather than negative as it implicitly indicates a longer survival time for the patients, which is also the endpoint of the study (survival endpoint). We note that the study seems to be enrolling some 5 patients per week, that the study has been enrolling since October 2006 and that patients are diagnosed with non-curable head & neck cancer with a median survival time of approx. 4 months when enrolled. 2/3s are receiving Zalutumumab and 1/3 are receiving best supportive care.
Solid performance to continue
Genmab has performed very well in 2008 with flat performance in the stock market. At the same time the Danish OMXC20 index, which Genmab is part of, is down by approx. 20%. We believe that the strong performance of Genmab is set to continue with the FDA BLA filing for Ofatumumab in CLL later this year and a potential approval and launch by mid 2009. Furthermore the stock could be boosted by strong data for Zalutumumab in head & neck cancer by Feb/Mar 2009 and a potential billion dollar deal for the compound afterwards. At our Science Session Dr. Baadsgaard did not comment on a potential deal size for Zalutumumab, but Genmab's CEO has at other occasions stated a deal a should be worth in excess of USD 1bn. A near-term trigger is the company's R&D day in Copenhagen on October 9, 2008. In terms of financials, Genmab has USD 168m or approx. DKK 1bn remaining from GSK in potential pre-approval milestones through 2009.
mfg ipollit
Genmab / Buy / DKK 308 - Solid impressions on Ofatumumab and EGFr from meeting with SVP Medical Affairs
• Very solid data on Ofatumumab in CLL - ASH presentation to be a strong trigger
• Delays to EGFr data confirmed to be a positive sign
• Good EGFr data could trigger next major deal
Looking forward to ASH in December
Handelsbanken Capital Markets hosted a "Science Session" with Genmab's SVP Medical Affairs Dr. Ole Baadsgaard aimed to discuss the Ofatumumab (anti CD20) programme, the phase III data released in chronic lymphocytic leukemia (CLL) as well as expectations for the phase III programme with Zalutumumab (anti EGFr) in head & neck cancer. There is no doubt in our mind that the phase III data with Ofatumumab in CLL are very strong and solid and that Genmab/GSK appear to be in possession of a very potent and effective antibody. Genmab has still not received confirmation that the data is accepted for presentation at ASH December 6-9 but the company expect data to be accepted. Furthermore we can sense on Dr. Baadsgaard that he would look very much forward to have the data presented at ASH due to the quality of data and strength of results. We believe that data will show that Ofatumumab also works in patients previously treated with Roche/Genentech's blockbuster antibody Rituxan and furthermore that there's a likelihood data could should that Ofatumumab has strong effect even in difficult genotypes with bad disease prognosis. We note that the data, according to Dr. Baadsgaard, surprised all participating parties in the Ofatumumab phase III study including the company as well as haematologists that had hoped for response rates of 30-35% and not the 44-51% as announced.
Looking forward to EGFr data
As argued above we look forward to ASH in December for the Ofatumumab phase III CLL data presentation. We look equally as much forward to the presentation of interim data/headline results for Genmab's EGFr antibody Zalutumumab in head & neck cancer. Dr. Baadsgaard confirmed at our meeting that the numerous delays to release of headline data from the phase III programme should be seen as positive rather than negative as it implicitly indicates a longer survival time for the patients, which is also the endpoint of the study (survival endpoint). We note that the study seems to be enrolling some 5 patients per week, that the study has been enrolling since October 2006 and that patients are diagnosed with non-curable head & neck cancer with a median survival time of approx. 4 months when enrolled. 2/3s are receiving Zalutumumab and 1/3 are receiving best supportive care.
Solid performance to continue
Genmab has performed very well in 2008 with flat performance in the stock market. At the same time the Danish OMXC20 index, which Genmab is part of, is down by approx. 20%. We believe that the strong performance of Genmab is set to continue with the FDA BLA filing for Ofatumumab in CLL later this year and a potential approval and launch by mid 2009. Furthermore the stock could be boosted by strong data for Zalutumumab in head & neck cancer by Feb/Mar 2009 and a potential billion dollar deal for the compound afterwards. At our Science Session Dr. Baadsgaard did not comment on a potential deal size for Zalutumumab, but Genmab's CEO has at other occasions stated a deal a should be worth in excess of USD 1bn. A near-term trigger is the company's R&D day in Copenhagen on October 9, 2008. In terms of financials, Genmab has USD 168m or approx. DKK 1bn remaining from GSK in potential pre-approval milestones through 2009.
mfg ipollit
etwas zu Isis und ViroPharma...
http://www.zacks.com/commentary/8659/Biotech+Names+with+Big+…
Biotech Names with Big Potential
With Jason Napodano
Sep 23, 2008
Today we continue our discussion with Zacks senior drug industry analyst Jason Napodano, CFA, who has been so kind as to run down his top Buy recommendations for us. We focus on biotech names in this second half of our discussion.
Now I know you have a Buy on Genentech – which has worked out quiet well by the way…
…Thanks for noticing!
And we’ve recently blogged positively on both Amgen and Biogen. Are those part of your blue-chip strategy, or is that the other direction you’re talking about?
Well, we still like Genentech (DNA). We think a higher bid from Roche is coming eventually. We’d hold onto DNA until that plays out. As for Amgen and Biogen (BIIB), we like both as core biotech holdings, but our alternative strategy for investors is more focused on smaller, mid-cap biotechs.
OK, so for a little more risk-tolerant investors?
Exactly! If you want big safe names, go with Abbott (ABT) and J&J (UJNJ). If you want a little more bang for your buck, and you can stomach some volatility, we’ve got two names for you.
The first is Isis Pharmaceuticals (ISIS). This is probably one of the best positioned biotech companies in world. It’s also our top-pick overall right now in healthcare. They are a leader in antisense technology.
Explain antisense for us, Jason…in the simplest terms, please!
Sure. Antisense drugs are short, chemically-modified complementary nucleotide chains that bind to a specific area of mRNA in order to inhibit translation (protein production). Basically, it’s a way to target disease at the genomic level. Instead of waiting for an errant gene to over-produce or under-produce a protein, enzyme or hormone, antisense drugs go directly after the genetic code. And so the possibilities are enormous because you are knocking out the cause of the disease instead of just treating the symptoms.
Isis’ technology is at the forefront of this revolutionary development platform. Isis has collaborations with the “who’s who” of healthcare – Merck (MRK), J&J, Eli Lilly (LLY), Novartis (NVS), Teva (TEVA) and Genzyme (GENZ). Because you are going after the gene, Isis can develop drugs over a wide scale of diseases.
The pipeline at Isis is enormous, and holds both proprietary and partnered compounds for cardiovascular disease, metabolic disorders, cancer, inflammatory diseases and infectious diseases. Take a look at the pipeline chart we have in our research report, it’s pretty impressive.
The leading candidate is in phase III trials for high cholesterol with partner Genzyme. This drug, mipomersen, has multi-billion dollar potential in our view. Other agents for diabetes, cancer and multiple sclerosis are also potential blockbusters. The pipeline fundamentals are particularly solid.
But the main thing we like about Isis is that the company is extremely well capitalized. Isis should exit 2008 with over $450 million in cash. Management should be able to raise another $190 million in 2009 by selling its Ibis biosensor business to Abbott Labs. That puts the company in an exceptionally solid financial situation to go along with the solid drug discovery / development fundamentals. Add the two together and you can see why it’s our top-pick right now.
Sounds like something investors should definitely look into. What’s the other idea you’ve got in mid-cap biotech?
It’s a name we’ve talked about in the past, Mark: ViroPharma (VPHM). This is one of our favorite names right now for several reasons. Firstly, management is among the best in the business. They’ve done a great job in building up ViroPharma through acquiring and in-licensing products over the past few years.
The leading product is Vancocin, a powerful antibiotic used to treat a life-threatening disease resulting from the infection of C. difficile. The story with Vancocin is that the drug was previously used only as a “last resort” for C. difficile infection (CDI), but recent clinical data, at a competitor nonetheless, shows that Vancocin is superior to the first-line therapy, call metronidazole, for severely infected patients.
So now management is out there spreading the world that Vancocin should be used in a greater percentage of patients other than just last resort cases. And they’re gaining traction because the IDSA (Infectious Disease Society of America) and SHEA (Society for Healthcare Epidemiology of America), two key scientific member organizations for infectious disease, are drafting guidelines that agree. So Vancocin continues to drive strong cash flow generation at ViroPharma.
And ViroPharma is profitable, if I remember correctly, right?
Correct. The second quarter 2008 was the 14th in a row of positive earnings. That’s an impressive feet for a mid-cap biotechnology company. But what all that strong cash flow generation has allowed ViroPharma management to do is build an impressive late-stage pipeline around Vancocin.
There are two candidates in late-stage development, maribavir for protection against cytomegalovirus (CMV) infection during stem cell and solid organ transplant, and Cinryze for hereditary angioedema (HAE). Maribavir is currently in two phase III trials with data expected in 2009. The phase II data on the drug has been very impressive, with patients showing 100% protection against CMV infection and a significant decline in transplant rejection, or something called graft vs. host disease (GvHD).
We think maribavir demonstrates a significant improvement over the current standard of care, a drug called ganciclovir made by Roche. Not only is it more efficacious, but the tolerability and safety profile is also superior. It could be a real breakthrough for patients undergoing stem cell transplant (SCT) or solid organ transplant (SOT). It’s also what’s known as an “orphan” indication, meaning ViroPharma will have strong pricing power and an exclusive market for at least seven years in the U.S. It’s a $500 million potential opportunity.
The second drug, Cinryze, also an “orphan” indication drug, is under regulatory review for HAE. HAE is a nasty inflammatory disease that results in significant swelling around the face, abdomen, hands and feet. The attacks, which could occur as often as 2-3 times per week, usually come without warning, and can be fatal if they occur inside the mouth or trachea. There’s a significant unmet medical need for a drug that can treat, or even prevent, these attacks. It’s another $500 million opportunity, in our view.
ViroPharma acquired the drug through the acquisition of Lev Pharmaceuticals earlier this year. The U.S. FDA is schedule to rule on the use of Cinryze as prophylactic (prevention) for HAE in October 2008. Given that an advisory panel previously recommended approval of the drug by a unanimous 21-0 margin in May 2008, we feel very good about the approval coming in October.
So by 2010 we think ViroPharma can have both maribavir and Cinryze, two orphan drugs each with $500 million potential, on the market. They’ve got an estimated $300 million in cash still even after the Lev Pharmaceuticals deal, and as of now, Vancocin remains patent protected.
Now that’s a potential risk with ViroPharma, correct? There is a patent challenge to Vancocin?
Yes, that’s the risk right now. But that risk has also created an opportunity to pick up ViroPharma on the cheap. A generic pharmaceutical company is looking to bring a bio-equivalent oral vancomycin to the market as soon as next year. However, ViroPharma has filed what’s known as a citizen’s petition, questioning the methods on which the generic company will look to prove bio-equivalence. It’s a strategy that has paid off well, having delayed the generic alternative since 2006 while the FDA’s Office of Generic Drug (OGD) looks into the issue.
It’s inevitable that a generic oral vancomycin will eventually come to market. Our financial model assumes it happens in 2010. You should know, that will cause a hit to earnings when it happens. But we think that investors should focus more on maribavir and Cinryze than when a generic Vancocin may come to market. The company should be able to maintain its strong history of profitability assuming both maribavir and Cinryze pan out. Given what we’ve seen so far, we like their chances.[/i]
mfg ipollit
http://www.zacks.com/commentary/8659/Biotech+Names+with+Big+…
Biotech Names with Big Potential
With Jason Napodano
Sep 23, 2008
Today we continue our discussion with Zacks senior drug industry analyst Jason Napodano, CFA, who has been so kind as to run down his top Buy recommendations for us. We focus on biotech names in this second half of our discussion.
Now I know you have a Buy on Genentech – which has worked out quiet well by the way…
…Thanks for noticing!
And we’ve recently blogged positively on both Amgen and Biogen. Are those part of your blue-chip strategy, or is that the other direction you’re talking about?
Well, we still like Genentech (DNA). We think a higher bid from Roche is coming eventually. We’d hold onto DNA until that plays out. As for Amgen and Biogen (BIIB), we like both as core biotech holdings, but our alternative strategy for investors is more focused on smaller, mid-cap biotechs.
OK, so for a little more risk-tolerant investors?
Exactly! If you want big safe names, go with Abbott (ABT) and J&J (UJNJ). If you want a little more bang for your buck, and you can stomach some volatility, we’ve got two names for you.
The first is Isis Pharmaceuticals (ISIS). This is probably one of the best positioned biotech companies in world. It’s also our top-pick overall right now in healthcare. They are a leader in antisense technology.
Explain antisense for us, Jason…in the simplest terms, please!
Sure. Antisense drugs are short, chemically-modified complementary nucleotide chains that bind to a specific area of mRNA in order to inhibit translation (protein production). Basically, it’s a way to target disease at the genomic level. Instead of waiting for an errant gene to over-produce or under-produce a protein, enzyme or hormone, antisense drugs go directly after the genetic code. And so the possibilities are enormous because you are knocking out the cause of the disease instead of just treating the symptoms.
Isis’ technology is at the forefront of this revolutionary development platform. Isis has collaborations with the “who’s who” of healthcare – Merck (MRK), J&J, Eli Lilly (LLY), Novartis (NVS), Teva (TEVA) and Genzyme (GENZ). Because you are going after the gene, Isis can develop drugs over a wide scale of diseases.
The pipeline at Isis is enormous, and holds both proprietary and partnered compounds for cardiovascular disease, metabolic disorders, cancer, inflammatory diseases and infectious diseases. Take a look at the pipeline chart we have in our research report, it’s pretty impressive.
The leading candidate is in phase III trials for high cholesterol with partner Genzyme. This drug, mipomersen, has multi-billion dollar potential in our view. Other agents for diabetes, cancer and multiple sclerosis are also potential blockbusters. The pipeline fundamentals are particularly solid.
But the main thing we like about Isis is that the company is extremely well capitalized. Isis should exit 2008 with over $450 million in cash. Management should be able to raise another $190 million in 2009 by selling its Ibis biosensor business to Abbott Labs. That puts the company in an exceptionally solid financial situation to go along with the solid drug discovery / development fundamentals. Add the two together and you can see why it’s our top-pick right now.
Sounds like something investors should definitely look into. What’s the other idea you’ve got in mid-cap biotech?
It’s a name we’ve talked about in the past, Mark: ViroPharma (VPHM). This is one of our favorite names right now for several reasons. Firstly, management is among the best in the business. They’ve done a great job in building up ViroPharma through acquiring and in-licensing products over the past few years.
The leading product is Vancocin, a powerful antibiotic used to treat a life-threatening disease resulting from the infection of C. difficile. The story with Vancocin is that the drug was previously used only as a “last resort” for C. difficile infection (CDI), but recent clinical data, at a competitor nonetheless, shows that Vancocin is superior to the first-line therapy, call metronidazole, for severely infected patients.
So now management is out there spreading the world that Vancocin should be used in a greater percentage of patients other than just last resort cases. And they’re gaining traction because the IDSA (Infectious Disease Society of America) and SHEA (Society for Healthcare Epidemiology of America), two key scientific member organizations for infectious disease, are drafting guidelines that agree. So Vancocin continues to drive strong cash flow generation at ViroPharma.
And ViroPharma is profitable, if I remember correctly, right?
Correct. The second quarter 2008 was the 14th in a row of positive earnings. That’s an impressive feet for a mid-cap biotechnology company. But what all that strong cash flow generation has allowed ViroPharma management to do is build an impressive late-stage pipeline around Vancocin.
There are two candidates in late-stage development, maribavir for protection against cytomegalovirus (CMV) infection during stem cell and solid organ transplant, and Cinryze for hereditary angioedema (HAE). Maribavir is currently in two phase III trials with data expected in 2009. The phase II data on the drug has been very impressive, with patients showing 100% protection against CMV infection and a significant decline in transplant rejection, or something called graft vs. host disease (GvHD).
We think maribavir demonstrates a significant improvement over the current standard of care, a drug called ganciclovir made by Roche. Not only is it more efficacious, but the tolerability and safety profile is also superior. It could be a real breakthrough for patients undergoing stem cell transplant (SCT) or solid organ transplant (SOT). It’s also what’s known as an “orphan” indication, meaning ViroPharma will have strong pricing power and an exclusive market for at least seven years in the U.S. It’s a $500 million potential opportunity.
The second drug, Cinryze, also an “orphan” indication drug, is under regulatory review for HAE. HAE is a nasty inflammatory disease that results in significant swelling around the face, abdomen, hands and feet. The attacks, which could occur as often as 2-3 times per week, usually come without warning, and can be fatal if they occur inside the mouth or trachea. There’s a significant unmet medical need for a drug that can treat, or even prevent, these attacks. It’s another $500 million opportunity, in our view.
ViroPharma acquired the drug through the acquisition of Lev Pharmaceuticals earlier this year. The U.S. FDA is schedule to rule on the use of Cinryze as prophylactic (prevention) for HAE in October 2008. Given that an advisory panel previously recommended approval of the drug by a unanimous 21-0 margin in May 2008, we feel very good about the approval coming in October.
So by 2010 we think ViroPharma can have both maribavir and Cinryze, two orphan drugs each with $500 million potential, on the market. They’ve got an estimated $300 million in cash still even after the Lev Pharmaceuticals deal, and as of now, Vancocin remains patent protected.
Now that’s a potential risk with ViroPharma, correct? There is a patent challenge to Vancocin?
Yes, that’s the risk right now. But that risk has also created an opportunity to pick up ViroPharma on the cheap. A generic pharmaceutical company is looking to bring a bio-equivalent oral vancomycin to the market as soon as next year. However, ViroPharma has filed what’s known as a citizen’s petition, questioning the methods on which the generic company will look to prove bio-equivalence. It’s a strategy that has paid off well, having delayed the generic alternative since 2006 while the FDA’s Office of Generic Drug (OGD) looks into the issue.
It’s inevitable that a generic oral vancomycin will eventually come to market. Our financial model assumes it happens in 2010. You should know, that will cause a hit to earnings when it happens. But we think that investors should focus more on maribavir and Cinryze than when a generic Vancocin may come to market. The company should be able to maintain its strong history of profitability assuming both maribavir and Cinryze pan out. Given what we’ve seen so far, we like their chances.[/i]
mfg ipollit
einige FDA-Entscheidungen in der nächsten Zeit (keine Ahnung wie vollständig)... von Relevanz hier nur Viropharma's Cinryze am 14.10.
Vielleicht noch interessant... AMAG mit Ferumoxytol kurz darauf.
http://seekingalpha.com/article/96751-fda-fall-decision-cale…

mfg ipollit
Vielleicht noch interessant... AMAG mit Ferumoxytol kurz darauf.
http://seekingalpha.com/article/96751-fda-fall-decision-cale…
mfg ipollit
Intercell...
23.09.2008
Intercell - s.aureus-Impfstoff ist laut Merrill für acht Euro je Aktie gut
Kaufempfehlung bestätigt - Chance von 25%, dass s.aureus die nötigen Effizienzschwellen erreicht
Merrill Lynch bestätigt die Kaufempfehlung für die Biotech-Aktie Intercell mit Kursziel 40 Euro. Analystin Erica Whittaker erwartet, dass Intercell und Partner Merck im ersten Halbjahr 2009 Zwischendaten von der Phase II/III-Studie zum s. aureus-Impfstoff V710 veröffentlichen. "Wir denken, es besteht ein signifikantes Risiko, dass V710 nicht die nötigen Effizienzschwellen erreicht, um einen deutlichen Schutz vor der Ansteckung mit s.aureus-Infektionen zu erreichen". In den Merrill-Berechnungen entfällt auf V710 allerdings weniger als 5% des Werts von Intercell. Auch sei in der aktuellen Bewertung ein möglicher Erfolg von V710 noch nicht eingepreist. Das führe dazu, dass V710 ein Bewertungspotenzial von bis zu 8 Euro je Aktie biete.
Experten, die sich mit s.aureus beschäftigen, seien zunehmend negativ für die Aussichten von V710 eingestellt, so die Analystin. Das habe mehrere Gründe: Zum einen sei bislang niemand mit der Entwicklung eines erfolgreichen Impfstoffes erfolgreich gewesen, auch sei es unwahrscheinlich, dass ein Impfstoff, der auf einem einzelnen Protein-Antigen basiert, Schutz vor diesem Krankheitserreger bietet.
Whittacker führt allerdings an, dass sie die bislang präsentierten vorklinischen Daten vielversprechend findet und sieht eine Chance von 25% für den Erfolg von V710. Auch Partner Merck sei hoffnungsfroh, laufen doch zwei grosse klinische Versuche.
Sollten diese Studien zeigen, dass V710 die Gefahr einer Infektion um mehr als 50% senken kann, dann dürfte dieser Impfstoff umfassend in exponierten Spitälern eingesetzt werden. Damit seien Umsätze von bis zu 1,8 Mrd. Dollar verbunden, so die Expertin. Intercell erhält von Merck zwischen 5% und 9% der V710-Umsätze, somit ergibt sich das Potenzial von bis zu 8 Euro je Aktie. Aktuell dürfte der Markt V710 lediglich mit 1,4 Euro je Aktie einpreisen.
Die von S. aureus ausgelösten Infektionen können zu Blutvergiftungen führen, aber auch zu Infektionen der Knochen, des Herzens und anderer innerer Organe, zu schwerwiegenden gesundheitlichen Problemen, dem Tod des Patienten und entsprechend zu erhöhten volkswirtschaftlichen Belastungen. Ausgangsmaterial für die Entwicklung des Impfstoffs ist ein von Intercell entdecktes und patentiertes Protein-Antigen. Dieses Antigen wurde 2004 mit Merck & Co. Inc. in eine weltweite exklusive Lizenzpartnerschaft eingebracht. (bs)
mfg ipollit
23.09.2008
Intercell - s.aureus-Impfstoff ist laut Merrill für acht Euro je Aktie gut
Kaufempfehlung bestätigt - Chance von 25%, dass s.aureus die nötigen Effizienzschwellen erreicht
Merrill Lynch bestätigt die Kaufempfehlung für die Biotech-Aktie Intercell mit Kursziel 40 Euro. Analystin Erica Whittaker erwartet, dass Intercell und Partner Merck im ersten Halbjahr 2009 Zwischendaten von der Phase II/III-Studie zum s. aureus-Impfstoff V710 veröffentlichen. "Wir denken, es besteht ein signifikantes Risiko, dass V710 nicht die nötigen Effizienzschwellen erreicht, um einen deutlichen Schutz vor der Ansteckung mit s.aureus-Infektionen zu erreichen". In den Merrill-Berechnungen entfällt auf V710 allerdings weniger als 5% des Werts von Intercell. Auch sei in der aktuellen Bewertung ein möglicher Erfolg von V710 noch nicht eingepreist. Das führe dazu, dass V710 ein Bewertungspotenzial von bis zu 8 Euro je Aktie biete.
Experten, die sich mit s.aureus beschäftigen, seien zunehmend negativ für die Aussichten von V710 eingestellt, so die Analystin. Das habe mehrere Gründe: Zum einen sei bislang niemand mit der Entwicklung eines erfolgreichen Impfstoffes erfolgreich gewesen, auch sei es unwahrscheinlich, dass ein Impfstoff, der auf einem einzelnen Protein-Antigen basiert, Schutz vor diesem Krankheitserreger bietet.
Whittacker führt allerdings an, dass sie die bislang präsentierten vorklinischen Daten vielversprechend findet und sieht eine Chance von 25% für den Erfolg von V710. Auch Partner Merck sei hoffnungsfroh, laufen doch zwei grosse klinische Versuche.
Sollten diese Studien zeigen, dass V710 die Gefahr einer Infektion um mehr als 50% senken kann, dann dürfte dieser Impfstoff umfassend in exponierten Spitälern eingesetzt werden. Damit seien Umsätze von bis zu 1,8 Mrd. Dollar verbunden, so die Expertin. Intercell erhält von Merck zwischen 5% und 9% der V710-Umsätze, somit ergibt sich das Potenzial von bis zu 8 Euro je Aktie. Aktuell dürfte der Markt V710 lediglich mit 1,4 Euro je Aktie einpreisen.
Die von S. aureus ausgelösten Infektionen können zu Blutvergiftungen führen, aber auch zu Infektionen der Knochen, des Herzens und anderer innerer Organe, zu schwerwiegenden gesundheitlichen Problemen, dem Tod des Patienten und entsprechend zu erhöhten volkswirtschaftlichen Belastungen. Ausgangsmaterial für die Entwicklung des Impfstoffs ist ein von Intercell entdecktes und patentiertes Protein-Antigen. Dieses Antigen wurde 2004 mit Merck & Co. Inc. in eine weltweite exklusive Lizenzpartnerschaft eingebracht. (bs)
mfg ipollit
OSIP... Tarceva kann zu tödlichen Leberschäden führen. Man muss allerdings hier auch das Verhältnis sehen... Tarveca wird bei sehr weit fortgeschrittenen Krebs (z.B. fortgeschrittener Lungenkrebs nach erfolgloser 1st-line und 2nd-line Therapie) angewendet, d.h. die Patienten sind so schon sehr schwer erkrankt, weshalb keine Therpaie wahrscheinlich ungleich mehr Schäden verursacht.
UPDATE 2-Tarceva makers warn of liver damage, 2 deaths
Tue Sep 23, 2008 6:31pm EDT
WASHINGTON, Sept 23 (Reuters) - Genentech Inc (DNA.N: Quote, Profile, Research, Stock Buzz) and OSI Pharmaceuticals (OSIP.O: Quote, Profile, Research, Stock Buzz) have alerted doctors about cases of liver damage among patients who took the cancer drug Tarceva in a post-approval study, U.S. regulators said on Tuesday.
One patient died from rapidly progressing liver failure and another died from a liver complication called hepatorenal syndrome, the companies said in a letter to doctors. The study was designed for patients with moderate liver impairment and advanced tumors.
The companies' letter was posted on the FDA's website.
"Patients with (liver) impairment should be monitored closely during therapy with Tarceva, and dosing should be interrupted or discontinued if changes in liver function are severe," the companies said in the letter.
Global sales of Tarceva hit $866 million in 2007. The drug's generic name is erlotinib.
Tarceva is an oral drug approved for treating certain patients with lung or pancreatic cancer. Most of the 36 patients in the post-approval study had types of tumors that are not approved for treatment with Tarceva, Genentech spokeswoman Charlotte Arnold said.
The prescribing information for Tarceva previously included a "precaution" about liver damage including fatalities. That language has been moved to the stronger "warnings" section of the drug's label, Arnold said.
Roche Holding AG (ROG.VX: Quote, Profile, Research, Stock Buzz) owns a majority stake in Genentech and has made an offer for the remaining shares. Genentech has rejected Roche's current offer of $89 a share. (Reporting by Lisa Richwine; Editing by Carol Bishopric and Bernard Orr)
mfg ipollit
UPDATE 2-Tarceva makers warn of liver damage, 2 deaths
Tue Sep 23, 2008 6:31pm EDT
WASHINGTON, Sept 23 (Reuters) - Genentech Inc (DNA.N: Quote, Profile, Research, Stock Buzz) and OSI Pharmaceuticals (OSIP.O: Quote, Profile, Research, Stock Buzz) have alerted doctors about cases of liver damage among patients who took the cancer drug Tarceva in a post-approval study, U.S. regulators said on Tuesday.
One patient died from rapidly progressing liver failure and another died from a liver complication called hepatorenal syndrome, the companies said in a letter to doctors. The study was designed for patients with moderate liver impairment and advanced tumors.
The companies' letter was posted on the FDA's website.
"Patients with (liver) impairment should be monitored closely during therapy with Tarceva, and dosing should be interrupted or discontinued if changes in liver function are severe," the companies said in the letter.
Global sales of Tarceva hit $866 million in 2007. The drug's generic name is erlotinib.
Tarceva is an oral drug approved for treating certain patients with lung or pancreatic cancer. Most of the 36 patients in the post-approval study had types of tumors that are not approved for treatment with Tarceva, Genentech spokeswoman Charlotte Arnold said.
The prescribing information for Tarceva previously included a "precaution" about liver damage including fatalities. That language has been moved to the stronger "warnings" section of the drug's label, Arnold said.
Roche Holding AG (ROG.VX: Quote, Profile, Research, Stock Buzz) owns a majority stake in Genentech and has made an offer for the remaining shares. Genentech has rejected Roche's current offer of $89 a share. (Reporting by Lisa Richwine; Editing by Carol Bishopric and Bernard Orr)
mfg ipollit
Antwort auf Beitrag Nr.: 35.243.589 von ipollit am 24.09.08 00:10:54Intercell... die Zulassung des JEV-Impfstoffes sollte schon weitestgehend eingepreist sein.
24.09.2008
Intercell - Wichtige Produktzulassung steht kurz bevor
Aktie dürfte zuletzt durch Abgaben seitens früherer Iomai-Aktionäre unter Druck gewesen sein
Die Anzeichen mehren sich, dass das Biotech-Unternehmen Intercell bereits in den nächsten Wochen seitens der US-Behörden die Zulassung für sein wichtigstes Produkt, den Impfstoff gegen Japanische Enzephalitis (JE), erhalten könnte. "Der Impfstoff findet sich auf dem Entwurf der Tagesordnung der Komitee-Sitzung des ACIP am 22./23. Oktober", schreibt Merrill Lynch-Analystin Erica Whittacker, die erst am Dienstag die Kaufempfehlung für Intercell mit Kursziel 40 Euro bestätigt hat.
Auffallend dabei: Dieses vom US Department of Health and Human Services eingesetzte Advisory Committee on Immunization Practices, das schriftliche Empfehlungen für die routinemässige Verab- reichung von Impfstoffen an Kinder und Erwachsene abgibt, beschäftigt sich normalerweise nur mit bereits zugelassenen Wirkstoffen. Damit liege die Vermutung nahe, dass das Komitee eine Zulassung für sehr wahrscheinlich hält, folgert Whittaker. Die Behandlung des JE-Impfstoffes ist für den frühen Nachmittag des 22.Oktober vorgesehen.
Auch Intercell-Vorstand Werner Lanthaler (im Bild) zeigt sich "sehr optimistisch", dass der JE-Impfstoff in den nächsten Wochen in den USA zugelassen wird. "Sie werden noch viel von uns hören", kündigte er am Dienstagabend bei der von Börse Express und Aktienforum veranstalteten Roadshow #8 in Graz an. So will Intercell mit Partner Merck in den nächsten sechs bis neun Monaten auch "signifikante Fortschritte" bezüglich des s.aureus-Impfstoffes V710 melden.
Zahlreiche Experten haben sich zuletzt kritisch geäussert, ob V710 die nötigen Schwellen erreicht, um einen deutlichen Schutz vor der Ansteckung mit hospitalen Infektionen zu gewährleisten. Ein Grund dafür ist u.a., dass bislang noch niemandem die Entwicklung eines solchen Impfstoffes geglückt ist. Merrill Lynch führt allerdings auch an, dass in der aktuellen Intercell-Bewertung ein möglicher Erfolg noch nicht eingepreist ist. Das führe dazu, dass V710 ein Bewertungspotenzial von bis zu 8 Euro je Aktie biete.
Intercell findet sich zudem unverändert auf der aktuellen Merrill Lynch-Liste der fünf "most preferred" Aktien im Bereich der Gesundheitsvorsorge in Europa. Merrill überarbeitet diese Liste monatlich, Intercell ist seit Ende März dieses Jahres darauf vertreten.
Im Kursverlauf der Intercell-Aktie macht sich dieser Optimismus aktuell noch nicht bemerkbar. Die Aktie hält sich im Vergleich zum Jahresbeginn zwar erstaunlich stabil, ist aber dennoch rund 20% von ihrem Jahreshoch bei knapp 33 Euro entfernt. Der Kursrückgang der vergangenen Tage - am Mittwoch tauchte die Aktie kurz unter die Marke von 26 Euro ab - dürfte vor allem Abgaben eines oder mehrerer Venture Capital-Fonds als Hintergrund haben, so ein Broker.
Diese VC-Fonds waren ursprünglich in die amerikanische Iomai investiert und haben im Zuge der Iomai-Übernahme durch Intercell Aktien des österreichischen Biotech-Unternehmens erhalten. Angesichts der starken Marktturbulenzen kommt es in privaten VC-Portfolios weltweit zu einem Exit-Druck. Betroffen sind davon dann in erster Linie jene Assets, die in diesen Fonds liquide sind - in diesem Fall die börsenotierte Intercell. Die fragliche Position sollte aber bereits grösstenteils abgebaut sein, ist zu hören. (bs)
mfg ipollit
24.09.2008
Intercell - Wichtige Produktzulassung steht kurz bevor
Aktie dürfte zuletzt durch Abgaben seitens früherer Iomai-Aktionäre unter Druck gewesen sein
Die Anzeichen mehren sich, dass das Biotech-Unternehmen Intercell bereits in den nächsten Wochen seitens der US-Behörden die Zulassung für sein wichtigstes Produkt, den Impfstoff gegen Japanische Enzephalitis (JE), erhalten könnte. "Der Impfstoff findet sich auf dem Entwurf der Tagesordnung der Komitee-Sitzung des ACIP am 22./23. Oktober", schreibt Merrill Lynch-Analystin Erica Whittacker, die erst am Dienstag die Kaufempfehlung für Intercell mit Kursziel 40 Euro bestätigt hat.
Auffallend dabei: Dieses vom US Department of Health and Human Services eingesetzte Advisory Committee on Immunization Practices, das schriftliche Empfehlungen für die routinemässige Verab- reichung von Impfstoffen an Kinder und Erwachsene abgibt, beschäftigt sich normalerweise nur mit bereits zugelassenen Wirkstoffen. Damit liege die Vermutung nahe, dass das Komitee eine Zulassung für sehr wahrscheinlich hält, folgert Whittaker. Die Behandlung des JE-Impfstoffes ist für den frühen Nachmittag des 22.Oktober vorgesehen.
Auch Intercell-Vorstand Werner Lanthaler (im Bild) zeigt sich "sehr optimistisch", dass der JE-Impfstoff in den nächsten Wochen in den USA zugelassen wird. "Sie werden noch viel von uns hören", kündigte er am Dienstagabend bei der von Börse Express und Aktienforum veranstalteten Roadshow #8 in Graz an. So will Intercell mit Partner Merck in den nächsten sechs bis neun Monaten auch "signifikante Fortschritte" bezüglich des s.aureus-Impfstoffes V710 melden.
Zahlreiche Experten haben sich zuletzt kritisch geäussert, ob V710 die nötigen Schwellen erreicht, um einen deutlichen Schutz vor der Ansteckung mit hospitalen Infektionen zu gewährleisten. Ein Grund dafür ist u.a., dass bislang noch niemandem die Entwicklung eines solchen Impfstoffes geglückt ist. Merrill Lynch führt allerdings auch an, dass in der aktuellen Intercell-Bewertung ein möglicher Erfolg noch nicht eingepreist ist. Das führe dazu, dass V710 ein Bewertungspotenzial von bis zu 8 Euro je Aktie biete.
Intercell findet sich zudem unverändert auf der aktuellen Merrill Lynch-Liste der fünf "most preferred" Aktien im Bereich der Gesundheitsvorsorge in Europa. Merrill überarbeitet diese Liste monatlich, Intercell ist seit Ende März dieses Jahres darauf vertreten.
Im Kursverlauf der Intercell-Aktie macht sich dieser Optimismus aktuell noch nicht bemerkbar. Die Aktie hält sich im Vergleich zum Jahresbeginn zwar erstaunlich stabil, ist aber dennoch rund 20% von ihrem Jahreshoch bei knapp 33 Euro entfernt. Der Kursrückgang der vergangenen Tage - am Mittwoch tauchte die Aktie kurz unter die Marke von 26 Euro ab - dürfte vor allem Abgaben eines oder mehrerer Venture Capital-Fonds als Hintergrund haben, so ein Broker.
Diese VC-Fonds waren ursprünglich in die amerikanische Iomai investiert und haben im Zuge der Iomai-Übernahme durch Intercell Aktien des österreichischen Biotech-Unternehmens erhalten. Angesichts der starken Marktturbulenzen kommt es in privaten VC-Portfolios weltweit zu einem Exit-Druck. Betroffen sind davon dann in erster Linie jene Assets, die in diesen Fonds liquide sind - in diesem Fall die börsenotierte Intercell. Die fragliche Position sollte aber bereits grösstenteils abgebaut sein, ist zu hören. (bs)
mfg ipollit
Antwort auf Beitrag Nr.: 35.242.712 von ipollit am 23.09.08 22:44:10noch zu AMAG... das Unternehmen sieht zwar nicht schlecht aus und das bei einer möglichen Zulassung von Ferumoxytol in den nächsten Wochen. Doch ist diese Zulassung anscheinend sehr unsicher... mir zu unsicher, um eine Position zu riskieren.
http://www.thestreet.com/_rms/s/amags-iron-drug-put-to-the-f…
AMAG's Iron Drug Put to the Fire
04/15/08 - 10:03 AM EDT
The ongoing bull-bear tug of war over AMAG Pharmaceuticals (AMAG Quote - Cramer on AMAG - Stock Picks) centers on one key question: Has the company studied enough patients using its intravenous iron therapy over a long enough time period to satisfy the safety requirements of U.S. regulators?
Lately, the bears have been winning this battle, especially since Merrill Lynch biotech analyst Andrew Berens jumped onto their side of the rope.
Berens' answer to the question above is an emphatic "no," which he spelled out when he downgraded AMAG from neutral to sell on April 9. Berens' negativity toward AMAG, which actually began in February, has helped push the stock down roughly 40% this year.
AMAG shares closed Monday at $35.57.
AMAG's intravenous iron therapy, known as ferumoxytol, can be administered in two, 17-second injections, far faster than conventional intravenous irons to treat anemia.
Ferumoxytol is being developed initially for use in patients with chronic kidney disease, including those on and off dialysis. AMAG completed four phase III studies and filed for approval in the U.S. last year. The Food and Drug Administration is expected to issue its decision by Oct. 19.
At Issue
Berens, citing discussions with FDA officials, claims that AMAG has taken shortcuts with the clinical development of ferumoxytol -- too few patients, too little follow-up, not enough dosing in "real world" situations to meet regulatory guidelines. Berens claims that AMAG is seeking approval of ferumoxytol as a "chronic use" treatment, which means that patients will need to be dosed repeatedly over time. As such, the FDA's safety standards are higher.
"We believe it unlikely that AMAG will be able to convince the FDA that the current development program accurately characterizes the safety profile of the drug as it is likely to be used in the 'real world' where dialysis patients receive 'repeated, intermittent use' of iron indefinitely," writes Berens in his April 9 note.
Berens further claims that AMAG's current database under review by the FDA only includes studies in which kidney disease patients received small doses of ferumoxytol for short periods of time.
The end result, says Berens: The FDA probably won't approve ferumoxytol this October, as the bulls expect. In fact, it's likely that before the October approval date arrives, the FDA will send a letter to AMAG instructing the company to run another year-long safety trial. This will push back ferumoxytol's launch in the U.S. by three years to 2011, he believes.
Is Berens right? Absolutely not, argues AMAG CEO Brian Pereira.
The Counter
To hear Periera explain it, ferumoxytol is not a chronic therapy in the strict definition of the term because kidney disease patients, whether they are on dialysis or not, will only receive ferumoxytol if and when their iron stores are depleted.
"What our [ferumoxytol filing] proposes is to treat iron-deficient anemia," says Pereira. Importantly, ferumoxytol will not be used to prevent iron-deficient anemia, he adds, which means that patients won't be dosed with the drug unless they need it to replenish iron in their system.
A regimen where ferumoxytol is administered to kidney disease patients quickly, only when their iron stores are depleted, is more an episodic treatment that truly mimics the real world setting in which these patients are treated, says Pereira. He rejects Berens' claim that giving repeated doses of ferumoxytol, whether patients need or not, is something the FDA requires.
Yes, some patients will receive repeat doses of ferumoxytol, but Periera says the company has data filed with the FDA to satisfy the agency's safety and exposure requirements.
"We worked closely with the FDA with respect to the design of our trials, the number of patients enrolled, the safety and efficacy database and the endpoints. How can he [Berens] have a better understanding of the agency's requirements than us," says Pereira.
The Takeaways
I'm the first one to say not to trust everything said by the CEO of a drug or biotech company. There are countless examples of investors led down the wrong path by management exaggerations or misstatements.
But in this case, I find it hard not to side with Pereira in the ferumoxytol argument. It's not that Berens doesn't present some good arguments. He does, and handicapping an FDA decision these days, especially one that seems to hinge on safety, is fraught with risk.
But when you speak with Pereira, or witness him answer questions about ferumoxytol in an investor meeting, he comes across as highly credible and assured. Moreover, this is not a guy learning the ins and outs of kidney disease and anemia on the job. Pereira is a highly trained physician and noted expert on kidney disease. He's a past president of the National Kidney Foundation and, literally, helped put together one of the seminal textbooks in the field.
So, in the Berens vs. Pereira fight, the edge, at least in my book, goes to Pereira.
There is no argument about ferumoxytol's efficacy, and the safety data presented so far at medical meetings comes up benign in terms of stuff to worry about from an FDA perspective. Intravenous iron has been a mainstay treatment for chronic kidney disease patients for years. Ferumoxytol is a much-needed improvement especially for non-dialysis patients, but it's still intravenous iron, which the FDA understands well.
Before the FDA issues its approval decision in October, look for the agency to possibly schedule an advisory committee meeting this summer to review the ferumoxytol data with a panel of kidney disease experts. This could be the first real view inside the FDA's thinking on the drug.
In the meantime, expect the bull-bear tug of war over ferumoxytol to continue.
mfg ipollit
http://www.thestreet.com/_rms/s/amags-iron-drug-put-to-the-f…
AMAG's Iron Drug Put to the Fire
04/15/08 - 10:03 AM EDT
The ongoing bull-bear tug of war over AMAG Pharmaceuticals (AMAG Quote - Cramer on AMAG - Stock Picks) centers on one key question: Has the company studied enough patients using its intravenous iron therapy over a long enough time period to satisfy the safety requirements of U.S. regulators?
Lately, the bears have been winning this battle, especially since Merrill Lynch biotech analyst Andrew Berens jumped onto their side of the rope.
Berens' answer to the question above is an emphatic "no," which he spelled out when he downgraded AMAG from neutral to sell on April 9. Berens' negativity toward AMAG, which actually began in February, has helped push the stock down roughly 40% this year.
AMAG shares closed Monday at $35.57.
AMAG's intravenous iron therapy, known as ferumoxytol, can be administered in two, 17-second injections, far faster than conventional intravenous irons to treat anemia.
Ferumoxytol is being developed initially for use in patients with chronic kidney disease, including those on and off dialysis. AMAG completed four phase III studies and filed for approval in the U.S. last year. The Food and Drug Administration is expected to issue its decision by Oct. 19.
At Issue
Berens, citing discussions with FDA officials, claims that AMAG has taken shortcuts with the clinical development of ferumoxytol -- too few patients, too little follow-up, not enough dosing in "real world" situations to meet regulatory guidelines. Berens claims that AMAG is seeking approval of ferumoxytol as a "chronic use" treatment, which means that patients will need to be dosed repeatedly over time. As such, the FDA's safety standards are higher.
"We believe it unlikely that AMAG will be able to convince the FDA that the current development program accurately characterizes the safety profile of the drug as it is likely to be used in the 'real world' where dialysis patients receive 'repeated, intermittent use' of iron indefinitely," writes Berens in his April 9 note.
Berens further claims that AMAG's current database under review by the FDA only includes studies in which kidney disease patients received small doses of ferumoxytol for short periods of time.
The end result, says Berens: The FDA probably won't approve ferumoxytol this October, as the bulls expect. In fact, it's likely that before the October approval date arrives, the FDA will send a letter to AMAG instructing the company to run another year-long safety trial. This will push back ferumoxytol's launch in the U.S. by three years to 2011, he believes.
Is Berens right? Absolutely not, argues AMAG CEO Brian Pereira.
The Counter
To hear Periera explain it, ferumoxytol is not a chronic therapy in the strict definition of the term because kidney disease patients, whether they are on dialysis or not, will only receive ferumoxytol if and when their iron stores are depleted.
"What our [ferumoxytol filing] proposes is to treat iron-deficient anemia," says Pereira. Importantly, ferumoxytol will not be used to prevent iron-deficient anemia, he adds, which means that patients won't be dosed with the drug unless they need it to replenish iron in their system.
A regimen where ferumoxytol is administered to kidney disease patients quickly, only when their iron stores are depleted, is more an episodic treatment that truly mimics the real world setting in which these patients are treated, says Pereira. He rejects Berens' claim that giving repeated doses of ferumoxytol, whether patients need or not, is something the FDA requires.
Yes, some patients will receive repeat doses of ferumoxytol, but Periera says the company has data filed with the FDA to satisfy the agency's safety and exposure requirements.
"We worked closely with the FDA with respect to the design of our trials, the number of patients enrolled, the safety and efficacy database and the endpoints. How can he [Berens] have a better understanding of the agency's requirements than us," says Pereira.
The Takeaways
I'm the first one to say not to trust everything said by the CEO of a drug or biotech company. There are countless examples of investors led down the wrong path by management exaggerations or misstatements.
But in this case, I find it hard not to side with Pereira in the ferumoxytol argument. It's not that Berens doesn't present some good arguments. He does, and handicapping an FDA decision these days, especially one that seems to hinge on safety, is fraught with risk.
But when you speak with Pereira, or witness him answer questions about ferumoxytol in an investor meeting, he comes across as highly credible and assured. Moreover, this is not a guy learning the ins and outs of kidney disease and anemia on the job. Pereira is a highly trained physician and noted expert on kidney disease. He's a past president of the National Kidney Foundation and, literally, helped put together one of the seminal textbooks in the field.
So, in the Berens vs. Pereira fight, the edge, at least in my book, goes to Pereira.
There is no argument about ferumoxytol's efficacy, and the safety data presented so far at medical meetings comes up benign in terms of stuff to worry about from an FDA perspective. Intravenous iron has been a mainstay treatment for chronic kidney disease patients for years. Ferumoxytol is a much-needed improvement especially for non-dialysis patients, but it's still intravenous iron, which the FDA understands well.
Before the FDA issues its approval decision in October, look for the agency to possibly schedule an advisory committee meeting this summer to review the ferumoxytol data with a panel of kidney disease experts. This could be the first real view inside the FDA's thinking on the drug.
In the meantime, expect the bull-bear tug of war over ferumoxytol to continue.
mfg ipollit
Micromet hat seine Cash-Reserven erfolgreich etwas aufgefüllt...
03.10.2008 13:05
Micromet Closes $40 Million Private Equity Placement
BETHESDA, Md., Oct. 3 /PRNewswire-FirstCall/ -- Micromet, (News) Inc. ("Micromet" or the "Company"), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced that it has closed its previously announced $40 million private placement financing. Participants include leading biotechnology investors Index Ventures Growth, Abingworth, DAFNA Capital Management, LLC, and Merlin Nexus, among others. Certain Micromet board members and affiliated entities also participated in the offering. The transaction was announced on September 30 and closed on October 2.
Micromet's President and CEO, Christian Itin stated: "We are very pleased to have completed this financing under these challenging market conditions, and we were gratified by the strong support of Micromet and the significant interest of the investors in this private placement. The proceeds from this financing will be used to advance our clinical antibody programs and to broaden our BiTE antibody pipeline."
Under the terms of the financing, Micromet sold 9.4 million shares of common stock and warrants to purchase 2.8 million shares of common stock at a total purchase price of $4.25 per unit, with each unit consisting of one share of common stock and a warrant to purchase 0.30 shares of common stock. The warrants are exercisable at $4.63 per share and will expire five years after the date of grant.
mfg ipollit
03.10.2008 13:05
Micromet Closes $40 Million Private Equity Placement
BETHESDA, Md., Oct. 3 /PRNewswire-FirstCall/ -- Micromet, (News) Inc. ("Micromet" or the "Company"), a biopharmaceutical company developing novel, proprietary antibodies for the treatment of cancer, inflammation and autoimmune diseases, today announced that it has closed its previously announced $40 million private placement financing. Participants include leading biotechnology investors Index Ventures Growth, Abingworth, DAFNA Capital Management, LLC, and Merlin Nexus, among others. Certain Micromet board members and affiliated entities also participated in the offering. The transaction was announced on September 30 and closed on October 2.
Micromet's President and CEO, Christian Itin stated: "We are very pleased to have completed this financing under these challenging market conditions, and we were gratified by the strong support of Micromet and the significant interest of the investors in this private placement. The proceeds from this financing will be used to advance our clinical antibody programs and to broaden our BiTE antibody pipeline."
Under the terms of the financing, Micromet sold 9.4 million shares of common stock and warrants to purchase 2.8 million shares of common stock at a total purchase price of $4.25 per unit, with each unit consisting of one share of common stock and a warrant to purchase 0.30 shares of common stock. The warrants are exercisable at $4.63 per share and will expire five years after the date of grant.
mfg ipollit
ein wenig etwas zur aktuellen Situation, die wahrlich nicht gut aussieht...
http://www.nytimes.com/2008/10/02/business/02crisis.html?_r=…
36 Hours of Alarm and Action as Crisis Spiraled
By JOE NOCERA
NYT October 2, 2008
This article was reported by Andrew Ross Sorkin, Diana B. Henriques, Edmund L. Andrews and Joe Nocera. It was written by Mr. Nocera.
“Panic can cause a prudent person to do rational things that can contribute to the failure of an institution.” — William A. Ackman of the hedge fund Pershing Square Capital Management.
It was early on Wednesday, Sept. 17, when executives at Pershing Square, Bill Ackman’s hedge fund, began getting nervous calls and e-mail messages from investors. Mr. Ackman, 42, has been a top Wall Street player for 15 years, making his clients — and himself — billions of dollars.
But now, Mr. Ackman and his colleagues were taken aback by what they were hearing. His big investors were worried about all of the Pershing assets held by Goldman Sachs, the blue-chip investment bank, whose stock had come under siege.
Never mind that Goldman kept Pershing’s assets in a segregated account, and that the money was safe. And never mind that Mr. Ackman believed Goldman was the world’s best-run investment bank and would come through the credit crisis unscathed.
Pershing investors still feared their money might be exposed. Mr. Ackman advised Goldman executives to do something to restore confidence — such as getting an infusion of capital from Warren E. Buffett, the billionaire investor. And while Mr. Ackman kept his assets at Goldman, he hurriedly set up accounts at three other institutions — just in case things got much worse.
Pershing had more faith than most. Up and down Wall Street, hedge funds with billions of dollars at Goldman and Morgan Stanley, another storied investment bank, were frantically pulling money out and looking for safer havens.
Panic was spreading on two of the scariest days ever in financial markets, and the biggest investors — not small investors — were panicking the most. Nobody was sure how much damage it would cause before it ended.
This is what a credit crisis looks like. It’s not like a stock market crisis, where the scary plunge of stocks is obvious to all. The credit crisis has played out in places most people can’t see. It’s banks refusing to lend to other banks — even though that is one of the most essential functions of the banking system. It’s a loss of confidence in seemingly healthy institutions like Morgan Stanley and Goldman — both of which reported profits even as the pressure was mounting. It is panicked hedge funds pulling out cash. It is frightened investors protecting themselves by buying credit-default swaps — a financial insurance policy against potential bankruptcy — at prices 30 times what they normally would pay.
It was this 36-hour period two weeks ago — from the morning of Wednesday, Sept. 17, to the afternoon of Thursday, Sept. 18 — that spooked policy makers by opening fissures in the worldwide financial system.
In their rush to do something, and do it fast, the Federal Reserve chairman, Ben S. Bernanke, and Treasury Secretary Henry M. Paulson Jr. concluded the time had come to use the “break the glass” rescue plan they had been developing. But in their urgency, they bypassed a crucial step in Washington and fashioned their $700 billion bailout without political spadework, which led to a resounding rejection this past Monday in the House of Representatives.
That Thursday evening, however, time was of the essence. In a hastily convened meeting in the conference room of the House speaker, Nancy Pelosi, the two men presented, in the starkest terms imaginable, the outline of the $700 billion plan to Congressional leaders. “If we don’t do this,” Mr. Bernanke said, according to several participants, “we may not have an economy on Monday.”
Setting the Stage
Wall Street executives and federal officials had known since the previous weekend that it was likely to be a difficult week.
With the government refusing to offer the same financial guarantees that helped save Bear Stearns, Fannie Mae and Freddie Mac, efforts on Saturday to find a buyer for Lehman Brothers had failed.
Sunday was spent preparing to deal with Lehman’s bankruptcy, which was announced Monday morning. Merrill Lynch, fearing it would be next, had agreed to be bought by Bank of America. The American International Group was near collapse. (It would be rescued with an $85 billion loan from the Federal Reserve on Tuesday evening.)
With government policy makers appearing to careen from crisis to crisis, the Dow Jones industrial average plunged 504 points on Monday, Sept. 15. Panic was in the air.
At those weekend meetings, Wall Street executives and federal officials talked about the possibility of contagion — that the Lehman bankruptcy might set off so much fear among investors that the market “would pivot to the next weakest firm in the herd,” as one federal official put it.
That firm, everyone knew, was likely to be Morgan Stanley, whose stock had been dropping since the previous Monday, Sept. 8. Within three hours on Tuesday, Sept. 16, Morgan Stanley shares fell another 28 percent, and the rising cost of its credit-default swaps suggested investors were predicting bankruptcy.
To allay the panic, the firm decided to report earnings a day early — after the market closed Tuesday afternoon instead of Wednesday morning. The profit was terrific — $1.425 billion, just a 3 percent decline from 2007 — and the thinking was that would give investors the night to absorb the good news.
“I am hoping that this will generally help calm the market,” Morgan Stanley’s chief financial officer, Colm A. Kelleher, said in an interview late that afternoon. “These markets are behaving irrationally. There’s a lot of fear.”
The Spreading Contagion
But contagion was already spreading. The problem posed by the Lehman bankruptcy was not the losses suffered by hedge funds and other investors who traded stocks or bonds with the firms. As federal officials had predicted, that turned out to be manageable. (That was one reason the government did not step in to save the firm.)
The real problem was that a handful of hedge funds that used the firm’s London office to handle their trades had billions of dollars in balances frozen in the bankruptcy.
Diamondback Capital Management, for instance, a $3 billion hedge fund, told its investors that 14.9 percent of its assets were locked up in the Lehman bankruptcy — money it could not extract. A number of other hedge funds were in the same predicament. (When called for comment, Diamondback officials did not respond.)
As this news spread, every other hedge fund manager had to worry about whether the balances they had at other Wall Street firms might suffer a similar fate. And Morgan Stanley and Goldman Sachs were the two biggest firms left that served this back-office role. That is why Mr. Ackman’s investors were calling him. And that is what caused hedge funds to pull money out of Morgan Stanley and Goldman Sachs, hedge their exposure by buying credit-default swaps that would cover losses if either firm couldn’t pay money they owed — or do both.
It was fear, not greed, that was driving everyone’s actions.
Breaking the Buck
There was another piece of bad news spooking investors — and government officials. On Tuesday, the Reserve Primary Fund, a $64 billion money market fund, and two smaller, related funds, revealed that they had “broken the buck” and would pay investors no more than 97 cents on the dollar.
Money market funds serve a critical role in greasing the wheels of commerce. They use investors’ money to make short-term loans, known as commercial paper, to big corporations like General Motors, I.B.M. and Microsoft. Commercial paper is attractive to money market funds because it pays them a higher interest rate than, say, United States Treasury bills, but is still considered relatively safe.
A run on money funds could force fund managers to shy away from commercial paper, fearing the loans were no longer safe. One reason given by the Reserve Primary Fund for breaking the buck was that it had bought Lehman commercial paper with a face value of $785 million that was now worth little because of its bankruptcy. If money market funds became fearful of buying commercial paper, that would make it far more difficult for companies to raise the cash needed to pay employees, for instance. At that point, it would not just be the credit markets that were frozen, but commerce itself.
Just as important, in the eyes of federal officials, was that money market funds had long been viewed by investors as akin to bank accounts — a safe place to store cash and earn interest on that money. Despite lacking federal deposit insurance, these funds held $3.4 trillion in assets.
“Breaking the buck was the Rubicon,” said a federal official. “This was the first time in the crisis that you could see stories talking about how it was affecting real people.”
Since that Monday, big institutional investors — like pension funds and college endowments — had been pulling money out of money funds. On Tuesday, individual investors joined the stampede.
At the Investment Company Institute, the trade group for the mutual fund industry, executives had organized a conference call that week with top-level fund executives and government officials.
“We were saying to Treasury and the Fed, at a very high level: Pay attention to this issue. This will have an impact,” recalled Greg Ahern, the group’s chief communication officer.
But government officials monitoring the crisis did not need the warning. They were already watching money fund outflows with alarm.
Surprisingly, stock investors — feeling better because of the government’s A.I.G. rescue plan — either did not comprehend or ignored the growing chaos in credit markets; the Dow actually rose 141.51 points on Tuesday.
A Dark Day
The respite was brief. Wednesday, Sept. 17, was one of those dark, ugly market days that offers not even a glimmer of hope.
Fearing the worst, Alex Ehrlich, the global head of prime services at the Swiss bank UBS, arrived at work in New York at 5 a.m. and immediately started putting out fires. Because he ran the firm’s prime brokerage unit, clients were calling to see whether their money was safe.
“We were being flooded with client requests to move positions, and the funding markets, which are critically important to prime brokers, were extremely volatile,” he said.
Within seconds of the market opening, the Dow was down 160 points. Among the big losers was Morgan Stanley. Despite the strong earnings it had disclosed late Tuesday, its stock continued to plummet. By noon, the Dow was down 330 points. It rallied in the afternoon, but went into free fall in the last 45 minutes, closing down 449 points.
And that was just what investors could see. Behind the scenes, the credit markets had almost completely frozen up. Banks were refusing to lend to other banks, and spreads on credit default swaps on financial stocks — the price of insuring against bankruptcy — veered into uncharted waters.
Moreover, the drain on money funds continued. By the end of business on Wednesday, institutional investors had withdrawn more than $290 billion from money market funds. In what experts call a “flight to safety,” investors were taking money out of stocks and bonds and even money market funds and buying the safest investments in the world: Treasury bills. As a result, yields on short-term Treasury bills dropped close to zero. That was almost unheard of.
In the stock market, Mr. Ehrlich of UBS was horrified by the plunge of Morgan Stanley’s shares, given the stellar earnings. “It felt like there was no ground beneath your feet,” he said. “I didn’t know where it was going to end.”
A Chief Executive’s Anger
Neither did Morgan Stanley’s chief executive, John J. Mack. A week before, his firm’s stock was trading in the mid-40s. On Wednesday, it fell from $28.70 a share to $21.75 — down about 50 percent over a week.
“There is no rational basis for the movements in our stock or credit default spreads,” Mr. Mack wrote in a companywide memo on Wednesday. Mr. Mack lashed out at the people he felt were responsible for Morgan Stanley’s woes: the short-sellers, who profit by betting that a stock will fall.
Like most Wall Street firms, Morgan Stanley over the years had handled transactions for short-sellers, despite complaints by other companies that short-sellers unfairly ganged up on their stock. Nevertheless, Mr. Mack called Senator Charles E. Schumer, Democrat of New York, and Christopher Cox, the chairman of the Securities and Exchange Commission, pressing them to ban short-selling.
He raged about what he viewed as a concerted effort to drive down the firm’s stock. “He got emotional,” says one person who knows him well.
Meeting with staff members Thursday morning as the stock plunged further — hitting a low of $11.70 midday — Mr. Mack said: “Listen. I know everybody is anxious about the stock price. I’m not selling any shares, and neither is my team. But I understand if you’re nervous and want to sell some shares.” Some did. (The company said fewer than one-third of employees sold stock that day.)
At the same time, Mr. Mack began talks to merge with Wachovia, and called other banks about possible combinations. He also called Mr. Buffett for advice, while aides in Tokyo contacted Mitsubishi UFJ, Japan’s biggest lender, hoping to raise additional capital.
Run on a Fund
Even as stocks tanked, turmoil was worsening in money markets. On Wednesday evening, Paul Schott Stevens, the head of the Investment Company Institute, learned about a problem with another money fund. “This time it was Putnam,” recalled Mr. Stevens, referring to the Boston-based mutual fund company Putnam Investments.
Out of the blue, it seemed, there was a run on the $12.3 billion Putnam Prime Money Market Fund. That meant the money fund contagion was spreading. Because of huge withdrawals, Putnam decided it had to shut the fund, and distribute the cash to shareholders. If it did not, the first ones out the door would get a better deal than the laggards.
Executives of the Investment Company Institute and fund officials scrambled to find a solution that would keep Putnam from having to take that step, but they failed. On Thursday, Putnam shuttered the fund. (After the government rescue plan was announced, it sold the fund, intact, to another company, and investors did not lose a penny.)
The Fed Takes Action
Ben Bernanke had spent his career studying financial crises. His first important work as an economist had been a study of the events that led to the Great Depression. Along with several economists, he came up with a phrase, “the financial accelerator,” which described how deteriorating market conditions could speed until they became unmanageable.
To an alarming degree, the credit crisis had played out as his academic work predicted. But his research also led Mr. Bernanke to the view that “situations where crises have really spiraled out of control are where the central bank has been on the sideline,” according to Mark Gertler, a New York University economist who has collaborated with Mr. Bernanke on some papers.
Mr. Bernanke had no intention of keeping the Fed on the sidelines. As the crisis deepened, it took more aggressive steps. It added liquidity to the system. It opened the discount window — the emergency lending facility that had been reserved for troubled banks — to investment banks. It also agreed to absorb up to $29 billion in Bear Stearns losses and made an $85 billion loan to keep A.I.G. afloat.
Representative Barney Frank, the Massachusetts Democrat who leads the House Financial Services Committee, asked Mr. Bernanke if the Fed had $85 billion to spare. “We have $800 billion,” Mr. Bernanke replied, according to Mr. Frank.
Since the Bear Stearns bailout, Treasury and Fed officials had discussed what a broad government intervention might look like. Although there were suggestions for a “bank holiday” — a temporary, nationwide closing of banks, which had not been done since 1933, to stem panicky withdrawals — Mr. Bernanke and Mr. Paulson dismissed the idea, fearing it would do far more harm than good by scaring people needlessly. They had both assembled teams to map out drastic rescue plans — the “break the glass” plans.
Almost from the start, they concluded the best systemic solution was to buy hard-to-sell mortgage-backed securities.
On Wednesday morning, during a conference call with other top officials, including Jean-Claude Trichet, the president of the European Central Bank, Mr. Bernanke sounded them out on a big government bailout. The other officials sounded relieved; their main questions were about whether Congress could act quickly.
That evening, Mr. Bernanke told Mr. Paulson during a conference call: “You have to go to Congress. This is pervasive.” Mr. Paulson agreed.
A Sense of Urgency
By Thursday morning, the need for dramatic action had grown even more urgent.
In Asia, stocks had already closed lower. To quell fears before the opening of European markets, the Fed and other central banks announced they would make $180 billion available, in an effort to get banks to start lending to each other again. The Fed had agreed to open its discount window to make loans available to money market funds to prevent further runs.
But it was to little avail.
At 8:30 Thursday morning in the United States, when Mr. Paulson and Mr. Bernanke reviewed the state of affairs, markets remained roiled. The crisis was not easing up.
One Bank’s Solution
Lloyd C. Blankfein, Goldman Sachs’s chief executive, had arrived at the firm’s office on 85 Broad Street just before 7 a.m. Thursday, anticipating another bad day. The investment bank’s stock had already been pummeled. From nearly $250 a share last October, it had fallen to $114.50 on Wednesday — after hitting a low of $97.78 that day.
One idea he had been exploring was to transform Goldman into a bank holding company. Mr. Mack, meantime, was also considering such a move for Morgan Stanley, and both were in separate discussions with the Fed. There was safety in that notion — they would become depository institutions regulated by the Fed and others — though it also meant they would not be able to pile on as much debt as they had as investment banks. That would hurt profits. But now profits were less pressing than survival. Mr. Blankfein accelerated the planning.
By 1 p.m., the Dow had fallen another 150 points — meaning that in a day and a half it was down nearly 600 points. Goldman’s stock dropped to $85.88, its lowest in nearly six years.
Just then, a prankster piped “The Star-Spangled Banner” over the firm’s loudspeaker system on the 50th floor. Fixed-income traders stopped and stood at attention, some with hands on their hearts. Oddly, it was at precisely that moment that the market — and Goldman’s shares — started to rise.
The traders began to cheer.
Curbing Short-Selling
What happened? At 1 p.m. New York time, the Financial Services Authority in Britain, which regulates that nation’s financial institutions, announced a ban on short-selling of 29 financial stocks that would last at least 30 days.
“When I saw that, I knew we were about to have the mother of all short squeezes,” said one hedge fund manager. Realizing that the S.E.C. was likely to follow suit, hedge funds began “covering their shorts” — that is, buying the stocks they had borrowed to short, even if it meant taking a loss.
That caused all kinds of stocks to begin rising. Sure enough, the S.E.C. followed suit the next day, placing a temporary short-selling ban on 799 financial stocks.
A few hours later came the second event. At 3:01 CNBC reported the Treasury and the Fed were planning a giant fund to buy toxic mortgage-backed assets from financial institutions. Though there had been hints of this earlier in the afternoon, and stocks had started rising around 2:30, the wide dissemination set off a huge rally. In a 45-minute burst, the Dow gained another 300 points, closing the day up 410 points.
Meeting on Capitol Hill
Two hours later, Mr. Paulson and Mr. Bernanke trooped up to Capitol Hill for a somber session with Congressional leaders. “That meeting was one of the most astounding experiences I’ve had in my 34 years in politics,” Senator Schumer recalled.
As the members of Congress and their aides listened, the two laid out their plan. They would begin offering federal insurance to money market funds immediately, in order to stop the run on money funds.
In addition, the S.E.C. would institute a ban on short-selling of financial stocks. Although Treasury officials concede that the move was mostly symbolic — investors can still buy put options that have the same effect as shorting stocks — they did it mainly “to scare the hell out of everybody,” as one official put it.
After Mr. Bernanke made his remark about the possibility that there might not be an economy on Monday without this plan, you could hear a pin drop.
“I gulped,” Mr. Schumer said.
Congressional leaders were nearly unanimous in saying that it needed to be done for the good of the country. Representative John A. Boehner of Ohio — the Republican House leader who a week later would lead the revolt against the plan — said it was time to put politics aside and move quickly, according to several participants. (An aide to Mr. Boehner denied that he voiced support for the plan, only that he made a plea for cooperation.)
Hearing that Mr. Bernanke and Mr. Paulson wanted legislation passed in a matter of days, the Senate majority leader, Harry Reid, expressed astonishment. “This is the United States Senate,” he said. “We can’t do it in that time frame.” His Republican counterpart, Senator Mitch McConnell, replied, “This time we can.”
He was wrong. After a week of wrangling, political infighting and compromise, the House on Monday voted down the legislation. The Dow plunged nearly 778 points, and credit markets had worsened, with interest rates rising and loans becoming harder to obtain.
Two weeks after Mr. Paulson and Mr. Bernanke made their appeal, the House is likely to try again.[/i]
mfg ipollit
http://www.nytimes.com/2008/10/02/business/02crisis.html?_r=…
36 Hours of Alarm and Action as Crisis Spiraled
By JOE NOCERA
NYT October 2, 2008
This article was reported by Andrew Ross Sorkin, Diana B. Henriques, Edmund L. Andrews and Joe Nocera. It was written by Mr. Nocera.
“Panic can cause a prudent person to do rational things that can contribute to the failure of an institution.” — William A. Ackman of the hedge fund Pershing Square Capital Management.
It was early on Wednesday, Sept. 17, when executives at Pershing Square, Bill Ackman’s hedge fund, began getting nervous calls and e-mail messages from investors. Mr. Ackman, 42, has been a top Wall Street player for 15 years, making his clients — and himself — billions of dollars.
But now, Mr. Ackman and his colleagues were taken aback by what they were hearing. His big investors were worried about all of the Pershing assets held by Goldman Sachs, the blue-chip investment bank, whose stock had come under siege.
Never mind that Goldman kept Pershing’s assets in a segregated account, and that the money was safe. And never mind that Mr. Ackman believed Goldman was the world’s best-run investment bank and would come through the credit crisis unscathed.
Pershing investors still feared their money might be exposed. Mr. Ackman advised Goldman executives to do something to restore confidence — such as getting an infusion of capital from Warren E. Buffett, the billionaire investor. And while Mr. Ackman kept his assets at Goldman, he hurriedly set up accounts at three other institutions — just in case things got much worse.
Pershing had more faith than most. Up and down Wall Street, hedge funds with billions of dollars at Goldman and Morgan Stanley, another storied investment bank, were frantically pulling money out and looking for safer havens.
Panic was spreading on two of the scariest days ever in financial markets, and the biggest investors — not small investors — were panicking the most. Nobody was sure how much damage it would cause before it ended.
This is what a credit crisis looks like. It’s not like a stock market crisis, where the scary plunge of stocks is obvious to all. The credit crisis has played out in places most people can’t see. It’s banks refusing to lend to other banks — even though that is one of the most essential functions of the banking system. It’s a loss of confidence in seemingly healthy institutions like Morgan Stanley and Goldman — both of which reported profits even as the pressure was mounting. It is panicked hedge funds pulling out cash. It is frightened investors protecting themselves by buying credit-default swaps — a financial insurance policy against potential bankruptcy — at prices 30 times what they normally would pay.
It was this 36-hour period two weeks ago — from the morning of Wednesday, Sept. 17, to the afternoon of Thursday, Sept. 18 — that spooked policy makers by opening fissures in the worldwide financial system.
In their rush to do something, and do it fast, the Federal Reserve chairman, Ben S. Bernanke, and Treasury Secretary Henry M. Paulson Jr. concluded the time had come to use the “break the glass” rescue plan they had been developing. But in their urgency, they bypassed a crucial step in Washington and fashioned their $700 billion bailout without political spadework, which led to a resounding rejection this past Monday in the House of Representatives.
That Thursday evening, however, time was of the essence. In a hastily convened meeting in the conference room of the House speaker, Nancy Pelosi, the two men presented, in the starkest terms imaginable, the outline of the $700 billion plan to Congressional leaders. “If we don’t do this,” Mr. Bernanke said, according to several participants, “we may not have an economy on Monday.”
Setting the Stage
Wall Street executives and federal officials had known since the previous weekend that it was likely to be a difficult week.
With the government refusing to offer the same financial guarantees that helped save Bear Stearns, Fannie Mae and Freddie Mac, efforts on Saturday to find a buyer for Lehman Brothers had failed.
Sunday was spent preparing to deal with Lehman’s bankruptcy, which was announced Monday morning. Merrill Lynch, fearing it would be next, had agreed to be bought by Bank of America. The American International Group was near collapse. (It would be rescued with an $85 billion loan from the Federal Reserve on Tuesday evening.)
With government policy makers appearing to careen from crisis to crisis, the Dow Jones industrial average plunged 504 points on Monday, Sept. 15. Panic was in the air.
At those weekend meetings, Wall Street executives and federal officials talked about the possibility of contagion — that the Lehman bankruptcy might set off so much fear among investors that the market “would pivot to the next weakest firm in the herd,” as one federal official put it.
That firm, everyone knew, was likely to be Morgan Stanley, whose stock had been dropping since the previous Monday, Sept. 8. Within three hours on Tuesday, Sept. 16, Morgan Stanley shares fell another 28 percent, and the rising cost of its credit-default swaps suggested investors were predicting bankruptcy.
To allay the panic, the firm decided to report earnings a day early — after the market closed Tuesday afternoon instead of Wednesday morning. The profit was terrific — $1.425 billion, just a 3 percent decline from 2007 — and the thinking was that would give investors the night to absorb the good news.
“I am hoping that this will generally help calm the market,” Morgan Stanley’s chief financial officer, Colm A. Kelleher, said in an interview late that afternoon. “These markets are behaving irrationally. There’s a lot of fear.”
The Spreading Contagion
But contagion was already spreading. The problem posed by the Lehman bankruptcy was not the losses suffered by hedge funds and other investors who traded stocks or bonds with the firms. As federal officials had predicted, that turned out to be manageable. (That was one reason the government did not step in to save the firm.)
The real problem was that a handful of hedge funds that used the firm’s London office to handle their trades had billions of dollars in balances frozen in the bankruptcy.
Diamondback Capital Management, for instance, a $3 billion hedge fund, told its investors that 14.9 percent of its assets were locked up in the Lehman bankruptcy — money it could not extract. A number of other hedge funds were in the same predicament. (When called for comment, Diamondback officials did not respond.)
As this news spread, every other hedge fund manager had to worry about whether the balances they had at other Wall Street firms might suffer a similar fate. And Morgan Stanley and Goldman Sachs were the two biggest firms left that served this back-office role. That is why Mr. Ackman’s investors were calling him. And that is what caused hedge funds to pull money out of Morgan Stanley and Goldman Sachs, hedge their exposure by buying credit-default swaps that would cover losses if either firm couldn’t pay money they owed — or do both.
It was fear, not greed, that was driving everyone’s actions.
Breaking the Buck
There was another piece of bad news spooking investors — and government officials. On Tuesday, the Reserve Primary Fund, a $64 billion money market fund, and two smaller, related funds, revealed that they had “broken the buck” and would pay investors no more than 97 cents on the dollar.
Money market funds serve a critical role in greasing the wheels of commerce. They use investors’ money to make short-term loans, known as commercial paper, to big corporations like General Motors, I.B.M. and Microsoft. Commercial paper is attractive to money market funds because it pays them a higher interest rate than, say, United States Treasury bills, but is still considered relatively safe.
A run on money funds could force fund managers to shy away from commercial paper, fearing the loans were no longer safe. One reason given by the Reserve Primary Fund for breaking the buck was that it had bought Lehman commercial paper with a face value of $785 million that was now worth little because of its bankruptcy. If money market funds became fearful of buying commercial paper, that would make it far more difficult for companies to raise the cash needed to pay employees, for instance. At that point, it would not just be the credit markets that were frozen, but commerce itself.
Just as important, in the eyes of federal officials, was that money market funds had long been viewed by investors as akin to bank accounts — a safe place to store cash and earn interest on that money. Despite lacking federal deposit insurance, these funds held $3.4 trillion in assets.
“Breaking the buck was the Rubicon,” said a federal official. “This was the first time in the crisis that you could see stories talking about how it was affecting real people.”
Since that Monday, big institutional investors — like pension funds and college endowments — had been pulling money out of money funds. On Tuesday, individual investors joined the stampede.
At the Investment Company Institute, the trade group for the mutual fund industry, executives had organized a conference call that week with top-level fund executives and government officials.
“We were saying to Treasury and the Fed, at a very high level: Pay attention to this issue. This will have an impact,” recalled Greg Ahern, the group’s chief communication officer.
But government officials monitoring the crisis did not need the warning. They were already watching money fund outflows with alarm.
Surprisingly, stock investors — feeling better because of the government’s A.I.G. rescue plan — either did not comprehend or ignored the growing chaos in credit markets; the Dow actually rose 141.51 points on Tuesday.
A Dark Day
The respite was brief. Wednesday, Sept. 17, was one of those dark, ugly market days that offers not even a glimmer of hope.
Fearing the worst, Alex Ehrlich, the global head of prime services at the Swiss bank UBS, arrived at work in New York at 5 a.m. and immediately started putting out fires. Because he ran the firm’s prime brokerage unit, clients were calling to see whether their money was safe.
“We were being flooded with client requests to move positions, and the funding markets, which are critically important to prime brokers, were extremely volatile,” he said.
Within seconds of the market opening, the Dow was down 160 points. Among the big losers was Morgan Stanley. Despite the strong earnings it had disclosed late Tuesday, its stock continued to plummet. By noon, the Dow was down 330 points. It rallied in the afternoon, but went into free fall in the last 45 minutes, closing down 449 points.
And that was just what investors could see. Behind the scenes, the credit markets had almost completely frozen up. Banks were refusing to lend to other banks, and spreads on credit default swaps on financial stocks — the price of insuring against bankruptcy — veered into uncharted waters.
Moreover, the drain on money funds continued. By the end of business on Wednesday, institutional investors had withdrawn more than $290 billion from money market funds. In what experts call a “flight to safety,” investors were taking money out of stocks and bonds and even money market funds and buying the safest investments in the world: Treasury bills. As a result, yields on short-term Treasury bills dropped close to zero. That was almost unheard of.
In the stock market, Mr. Ehrlich of UBS was horrified by the plunge of Morgan Stanley’s shares, given the stellar earnings. “It felt like there was no ground beneath your feet,” he said. “I didn’t know where it was going to end.”
A Chief Executive’s Anger
Neither did Morgan Stanley’s chief executive, John J. Mack. A week before, his firm’s stock was trading in the mid-40s. On Wednesday, it fell from $28.70 a share to $21.75 — down about 50 percent over a week.
“There is no rational basis for the movements in our stock or credit default spreads,” Mr. Mack wrote in a companywide memo on Wednesday. Mr. Mack lashed out at the people he felt were responsible for Morgan Stanley’s woes: the short-sellers, who profit by betting that a stock will fall.
Like most Wall Street firms, Morgan Stanley over the years had handled transactions for short-sellers, despite complaints by other companies that short-sellers unfairly ganged up on their stock. Nevertheless, Mr. Mack called Senator Charles E. Schumer, Democrat of New York, and Christopher Cox, the chairman of the Securities and Exchange Commission, pressing them to ban short-selling.
He raged about what he viewed as a concerted effort to drive down the firm’s stock. “He got emotional,” says one person who knows him well.
Meeting with staff members Thursday morning as the stock plunged further — hitting a low of $11.70 midday — Mr. Mack said: “Listen. I know everybody is anxious about the stock price. I’m not selling any shares, and neither is my team. But I understand if you’re nervous and want to sell some shares.” Some did. (The company said fewer than one-third of employees sold stock that day.)
At the same time, Mr. Mack began talks to merge with Wachovia, and called other banks about possible combinations. He also called Mr. Buffett for advice, while aides in Tokyo contacted Mitsubishi UFJ, Japan’s biggest lender, hoping to raise additional capital.
Run on a Fund
Even as stocks tanked, turmoil was worsening in money markets. On Wednesday evening, Paul Schott Stevens, the head of the Investment Company Institute, learned about a problem with another money fund. “This time it was Putnam,” recalled Mr. Stevens, referring to the Boston-based mutual fund company Putnam Investments.
Out of the blue, it seemed, there was a run on the $12.3 billion Putnam Prime Money Market Fund. That meant the money fund contagion was spreading. Because of huge withdrawals, Putnam decided it had to shut the fund, and distribute the cash to shareholders. If it did not, the first ones out the door would get a better deal than the laggards.
Executives of the Investment Company Institute and fund officials scrambled to find a solution that would keep Putnam from having to take that step, but they failed. On Thursday, Putnam shuttered the fund. (After the government rescue plan was announced, it sold the fund, intact, to another company, and investors did not lose a penny.)
The Fed Takes Action
Ben Bernanke had spent his career studying financial crises. His first important work as an economist had been a study of the events that led to the Great Depression. Along with several economists, he came up with a phrase, “the financial accelerator,” which described how deteriorating market conditions could speed until they became unmanageable.
To an alarming degree, the credit crisis had played out as his academic work predicted. But his research also led Mr. Bernanke to the view that “situations where crises have really spiraled out of control are where the central bank has been on the sideline,” according to Mark Gertler, a New York University economist who has collaborated with Mr. Bernanke on some papers.
Mr. Bernanke had no intention of keeping the Fed on the sidelines. As the crisis deepened, it took more aggressive steps. It added liquidity to the system. It opened the discount window — the emergency lending facility that had been reserved for troubled banks — to investment banks. It also agreed to absorb up to $29 billion in Bear Stearns losses and made an $85 billion loan to keep A.I.G. afloat.
Representative Barney Frank, the Massachusetts Democrat who leads the House Financial Services Committee, asked Mr. Bernanke if the Fed had $85 billion to spare. “We have $800 billion,” Mr. Bernanke replied, according to Mr. Frank.
Since the Bear Stearns bailout, Treasury and Fed officials had discussed what a broad government intervention might look like. Although there were suggestions for a “bank holiday” — a temporary, nationwide closing of banks, which had not been done since 1933, to stem panicky withdrawals — Mr. Bernanke and Mr. Paulson dismissed the idea, fearing it would do far more harm than good by scaring people needlessly. They had both assembled teams to map out drastic rescue plans — the “break the glass” plans.
Almost from the start, they concluded the best systemic solution was to buy hard-to-sell mortgage-backed securities.
On Wednesday morning, during a conference call with other top officials, including Jean-Claude Trichet, the president of the European Central Bank, Mr. Bernanke sounded them out on a big government bailout. The other officials sounded relieved; their main questions were about whether Congress could act quickly.
That evening, Mr. Bernanke told Mr. Paulson during a conference call: “You have to go to Congress. This is pervasive.” Mr. Paulson agreed.
A Sense of Urgency
By Thursday morning, the need for dramatic action had grown even more urgent.
In Asia, stocks had already closed lower. To quell fears before the opening of European markets, the Fed and other central banks announced they would make $180 billion available, in an effort to get banks to start lending to each other again. The Fed had agreed to open its discount window to make loans available to money market funds to prevent further runs.
But it was to little avail.
At 8:30 Thursday morning in the United States, when Mr. Paulson and Mr. Bernanke reviewed the state of affairs, markets remained roiled. The crisis was not easing up.
One Bank’s Solution
Lloyd C. Blankfein, Goldman Sachs’s chief executive, had arrived at the firm’s office on 85 Broad Street just before 7 a.m. Thursday, anticipating another bad day. The investment bank’s stock had already been pummeled. From nearly $250 a share last October, it had fallen to $114.50 on Wednesday — after hitting a low of $97.78 that day.
One idea he had been exploring was to transform Goldman into a bank holding company. Mr. Mack, meantime, was also considering such a move for Morgan Stanley, and both were in separate discussions with the Fed. There was safety in that notion — they would become depository institutions regulated by the Fed and others — though it also meant they would not be able to pile on as much debt as they had as investment banks. That would hurt profits. But now profits were less pressing than survival. Mr. Blankfein accelerated the planning.
By 1 p.m., the Dow had fallen another 150 points — meaning that in a day and a half it was down nearly 600 points. Goldman’s stock dropped to $85.88, its lowest in nearly six years.
Just then, a prankster piped “The Star-Spangled Banner” over the firm’s loudspeaker system on the 50th floor. Fixed-income traders stopped and stood at attention, some with hands on their hearts. Oddly, it was at precisely that moment that the market — and Goldman’s shares — started to rise.
The traders began to cheer.
Curbing Short-Selling
What happened? At 1 p.m. New York time, the Financial Services Authority in Britain, which regulates that nation’s financial institutions, announced a ban on short-selling of 29 financial stocks that would last at least 30 days.
“When I saw that, I knew we were about to have the mother of all short squeezes,” said one hedge fund manager. Realizing that the S.E.C. was likely to follow suit, hedge funds began “covering their shorts” — that is, buying the stocks they had borrowed to short, even if it meant taking a loss.
That caused all kinds of stocks to begin rising. Sure enough, the S.E.C. followed suit the next day, placing a temporary short-selling ban on 799 financial stocks.
A few hours later came the second event. At 3:01 CNBC reported the Treasury and the Fed were planning a giant fund to buy toxic mortgage-backed assets from financial institutions. Though there had been hints of this earlier in the afternoon, and stocks had started rising around 2:30, the wide dissemination set off a huge rally. In a 45-minute burst, the Dow gained another 300 points, closing the day up 410 points.
Meeting on Capitol Hill
Two hours later, Mr. Paulson and Mr. Bernanke trooped up to Capitol Hill for a somber session with Congressional leaders. “That meeting was one of the most astounding experiences I’ve had in my 34 years in politics,” Senator Schumer recalled.
As the members of Congress and their aides listened, the two laid out their plan. They would begin offering federal insurance to money market funds immediately, in order to stop the run on money funds.
In addition, the S.E.C. would institute a ban on short-selling of financial stocks. Although Treasury officials concede that the move was mostly symbolic — investors can still buy put options that have the same effect as shorting stocks — they did it mainly “to scare the hell out of everybody,” as one official put it.
After Mr. Bernanke made his remark about the possibility that there might not be an economy on Monday without this plan, you could hear a pin drop.
“I gulped,” Mr. Schumer said.
Congressional leaders were nearly unanimous in saying that it needed to be done for the good of the country. Representative John A. Boehner of Ohio — the Republican House leader who a week later would lead the revolt against the plan — said it was time to put politics aside and move quickly, according to several participants. (An aide to Mr. Boehner denied that he voiced support for the plan, only that he made a plea for cooperation.)
Hearing that Mr. Bernanke and Mr. Paulson wanted legislation passed in a matter of days, the Senate majority leader, Harry Reid, expressed astonishment. “This is the United States Senate,” he said. “We can’t do it in that time frame.” His Republican counterpart, Senator Mitch McConnell, replied, “This time we can.”
He was wrong. After a week of wrangling, political infighting and compromise, the House on Monday voted down the legislation. The Dow plunged nearly 778 points, and credit markets had worsened, with interest rates rising and loans becoming harder to obtain.
Two weeks after Mr. Paulson and Mr. Bernanke made their appeal, the House is likely to try again.[/i]
mfg ipollit
Antwort auf Beitrag Nr.: 35.395.521 von ipollit am 03.10.08 13:17:371 Trillion = 1 Billionen = 1000 Milliarden...
http://money.cnn.com/2008/09/30/magazines/fortune/varchaver_…
The $55 trillion question
The financial crisis has put a spotlight on the obscure world of credit default swaps - which trade in a vast, unregulated market that most people haven't heard of and even fewer understand. Will this be the next disaster?
By Nicholas Varchaver, senior editor and Katie Benner, writer-reporter
Last Updated: September 30, 2008: 12:28 PM ET
(Fortune Magazine) -- As Congress wrestles with another bailout bill to try to contain the financial contagion, there's a potential killer bug out there whose next movement can't be predicted: the Credit Default Swap.
In just over a decade these privately traded derivatives contracts have ballooned from nothing into a $54.6 trillion market. CDS are the fastest-growing major type of financial derivatives. More important, they've played a critical role in the unfolding financial crisis. First, by ostensibly providing "insurance" on risky mortgage bonds, they encouraged and enabled reckless behavior during the housing bubble.
"If CDS had been taken out of play, companies would've said, 'I can't get this [risk] off my books,'" says Michael Greenberger, a University of Maryland law professor and former director of trading and markets at the Commodity Futures Trading Commission. "If they couldn't keep passing the risk down the line, those guys would've been stopped in their tracks. The ultimate assurance for issuing all this stuff was, 'It's insured.'"
Second, terror at the potential for a financial Ebola virus radiating out from a failing institution and infecting dozens or hundreds of other companies - all linked to one another by CDS and other instruments - was a major reason that regulators stepped in to bail out Bear Stearns and buy out AIG (AIG, Fortune 500), whose calamitous descent itself was triggered by losses on its CDS contracts (see "Hank's Last Stand").
And the fear of a CDS catastrophe still haunts the markets. For starters, nobody knows how federal intervention might ripple through this chain of contracts. And meanwhile, as we'll see, two fundamental aspects of the CDS market - that it is unregulated, and that almost nothing is disclosed publicly - may be about to change. That adds even more uncertainty to the equation.
"The big problem is that here are all these public companies - banks and corporations - and no one really knows what exposure they've got from the CDS contracts," says Frank Partnoy, a law professor at the University of San Diego and former Morgan Stanley derivatives salesman who has been writing about the dangers of CDS and their ilk for a decade. "The really scary part is that we don't have a clue." Chris Wolf, a co-manager of Cogo Wolf, a hedge fund of funds, compares them to one of the great mysteries of astrophysics: "This has become essentially the dark matter of the financial universe."
***
AT FIRST GLANCE, credit default swaps don't look all that scary. A CDS is just a contract: The "buyer" plunks down something that resembles a premium, and the "seller" agrees to make a specific payment if a particular event, such as a bond default, occurs. Used soberly, CDS offer concrete benefits: If you're holding bonds and you're worried that the issuer won't be able to pay, buying CDS should cover your loss. "CDS serve a very useful function of allowing financial markets to efficiently transfer credit risk," argues Sunil Hirani, the CEO of Creditex, one of a handful of marketplaces that trade the contracts.
Because they're contracts rather than securities or insurance, CDS are easy to create: Often deals are done in a one-minute phone conversation or an instant message. Many technical aspects of CDS, such as the typical five-year term, have been standardized by the International Swaps and Derivatives Association (ISDA). That only accelerates the process. You strike your deal, fill out some forms, and you've got yourself a $5 million - or a $100 million - contract.
And as long as someone is willing to take the other side of the proposition, a CDS can cover just about anything, making it the Wall Street equivalent of those notorious Lloyds of London policies covering Liberace's hands and other esoterica. It has even become possible to purchase a CDS that would pay out if the U.S. government defaults. (Trust us when we say that if the government goes under, trying to collect will be the least of your worries.)
You can guess how Wall Street cowboys responded to the opportunity to make deals that (1) can be struck in a minute, (2) require little or no cash upfront, and (3) can cover anything. Yee-haw! You can almost picture Slim Pickens in Dr. Strangelove climbing onto the H-bomb before it's released from the B-52. And indeed, the volume of CDS has exploded with nuclear force, nearly doubling every year since 2001 to reach a recent peak of $62 trillion at the end of 2007, before receding to $54.6 trillion as of June 30, according to ISDA.
Take that gargantuan number with a grain of salt. It refers to the face value of all outstanding contracts. But many players in the market hold offsetting positions. So if, in theory, every entity that owns CDS had to settle its contracts tomorrow and "netted" all its positions against each other, a much smaller amount of money would change hands. But even a tiny fraction of that $54.6 trillion would still be a daunting sum.
The credit freeze and then the Bear disaster explain the drop in outstanding CDS contracts during the first half of the year - and the market has only worsened since. CDS contracts on widely held debt, such as General Motors' (GM, Fortune 500), continue to be actively bought and sold. But traders say almost no new contracts are being written on any but the most liquid debt issues right now, in part because nobody wants to put money at risk and because nobody knows what Washington will do and how that will affect the market. ("There's nothing to do but watch Bernanke on TV," one trader told Fortune during the week when the Fed chairman was going before Congress to push the mortgage bailout.) So, after nearly a decade of exponential growth, the CDS market is poised for its first sustained contraction.
***
ONE REASON THE MARKET TOOK OFF is that you don't have to own a bond to buy a CDS on it - anyone can place a bet on whether a bond will fail. Indeed the majority of CDS now consists of bets on other people's debt. That's why it's possible for the market to be so big: The $54.6 trillion in CDS contracts completely dwarfs total corporate debt, which the Securities Industry and Financial Markets Association puts at $6.2 trillion, and the $10 trillion it counts in all forms of asset-backed debt.
"It's sort of like I think you're a bad driver and you're going to crash your car," says Greenberger, formerly of the CFTC. "So I go to an insurance company and get collision insurance on your car because I think it'll crash and I'll collect on it." That's precisely what the biggest winners in the subprime debacle did. Hedge fund star John Paulson of Paulson & Co., for example, made $15 billion in 2007, largely by using CDS to bet that other investors' subprime mortgage bonds would default.
So what started out as a vehicle for hedging ended up giving investors a cheap, easy way to wager on almost any event in the credit markets. In effect, credit default swaps became the world's largest casino. As Christopher Whalen, a managing director of Institutional Risk Analytics, observes, "To be generous, you could call it an unregulated, uncapitalized insurance market. But really, you would call it a gaming contract."
There is at least one key difference between casino gambling and CDS trading: Gambling has strict government regulation. The federal government has long shied away from any oversight of CDS. The CFTC floated the idea of taking an oversight role in the late '90s, only to find itself opposed by Federal Reserve chairman Alan Greenspan and others. Then, in 2000, Congress, with the support of Greenspan and Treasury Secretary Lawrence Summers, passed a bill prohibiting all federal and most state regulation of CDS and other derivatives. In a press release at the time, co-sponsor Senator Phil Gramm - most recently in the news when he stepped down as John McCain's campaign co-chair this summer after calling people who talk about a recession "whiners" - crowed that the new law "protects financial institutions from over-regulation ... and it guarantees that the United States will maintain its global dominance of financial markets." (The authors of the legislation were so bent on warding off regulation that they had the bill specify that it would "supersede and preempt the application of any state or local law that prohibits gaming ...") Not everyone was as sanguine as Gramm. In 2003 Warren Buffett famously called derivatives "financial weapons of mass destruction."
***
THERE'S ANOTHER BIG difference between trading CDS and casino gambling. When you put $10 on black 22, you're pretty sure the casino will pay off if you win. The CDS market offers no such assurance. One reason the market grew so quickly was that hedge funds poured in, sensing easy money. And not just big, well-established hedge funds but a lot of upstarts. So in some cases, giant financial institutions were counting on collecting money from institutions only slightly more solvent than your average minimart. The danger, of course, is that if a hedge fund suddenly has to pay off on a lot of CDS, it will simply go out of business. "People have been insuring risks that they can't insure," says Peter Schiff, the president of Euro Pacific Capital and author of Crash Proof, which predicted doom for Fannie and Freddie, among other things. "Let's say you're writing fire insurance policies, and every time you get the [premium], you spend it. You just assume that no houses are going to burn down. And all of a sudden there's a huge fire and they all burn down. What do you do? You just close up shop."


This is not an academic concern. Wachovia (WB, Fortune 500) and Citigroup (C, Fortune 500) are wrangling in court with a $50 million hedge fund located in the Channel Islands. The reason: A dispute over two $10 million credit default swaps covering some CDOs. The specifics of the spat aren't important. What's most revealing is that these massive banks put their faith in a Lilliputian fund (in an inaccessible jurisdiction) that was risking 40% of its capital for just two CDS. Can anyone imagine that Citi would, say, insure its headquarters building with a thinly capitalized, unregulated, offshore entity?
That's one element of what's known as "counterparty risk." Here's another: In many cases, you don't even know who has the other side of your bet. Parties to the contract can, and do, transfer their side of the contract to third parties. Investment firms assert that transfers are well documented (a claim that, like most in the world of CDS, is impossible to verify). But even if that's true, you're still left with the fact that a given company's risks are being dispersed in ways that they may not know about and can't control.
It doesn't help that CDS trading is a haphazard process. Most contracts are bought and sold over the phone or by instant message and settled manually. Settlement has been sloppy, confirms Jamie Cawley of IDX Capital, a firm that brokers trades between big banks. Pushed by New York Fed president Timothy Geithner, the players have been improving the process. But even as recently as a year ago, Cawley says, so many trades were sitting around unfulfilled that "there were $1 trillion worth of swaps that were unsettled among counterparties."
Trade settlement is not the only anachronistic aspect of CDS trading. Consider what will happen with CDS contracts relating to Fannie Mae and Freddie Mac. The two were placed in conservatorship on Sept. 7. But the value of many contracts won't be determined till Oct. 6, when an auction will set a cash price for Fannie and Freddie bonds. We'll spare you the technical reasons, but suffice it to ask: Can you imagine any other major market that would need a month to resolve something like this?
***
WITH WASHINGTON SUDDENLY in a frenzy of outrage over the financial markets, debating everything from the shape and extent of the mortgage plan to what should be done about short-selling, the future for CDS is very blurry. "The market is here to stay," asserts Cawley. The question is simply: What sorts of changes are in store? As this article was going to press, SEC chairman Christopher Cox asked the Senate to allow his agency to begin regulating CDS - mostly, it should be said, to rein in short-selling. And the SEC separately announced that it was expanding its investigation of market manipulation, which initially targeted the short-sellers, to CDS investors.
Under other circumstances, Cox's request might have been met with polite silence. But the convulsions over the mortgage bailout are so dramatic that they are reminiscent of the moment, soon after the Enron scandal, when Congress drafted the Sarbanes-Oxley legislation. The desire to blame short-sellers may actually result in powers for Cox that, until very recently, he showed no signs of wanting. Should legislators wade into this issue, the measures most widely seen as necessary are straightforward: some form of centralized trading or clearing and some form of capital or reserve requirements. Meanwhile, New York State's insurance commissioner, Eric Dinallo, announced new regulations that would essentially treat sellers of some (but not all) CDS as insurance entities, thereby forcing them to set aside reserves and otherwise follow state insurance law - requirements that would probably drive many participants from the market. Whether CDS players will find a way to challenge the rules remains to be seen. (ISDA, the industry's trade group, has already gone on record in opposition to Cox's proposal.) If nothing else, the New York law may provide additional impetus for the feds to take action.
For now, the biggest impact could come from the Financial Accounting Standards Board. It is implementing a new rule in November that will require sellers of CDS and other credit derivatives to report detailed information, including their maximum payouts and reasons for entering the contracts, as well as assets that might allow them to offset any payouts. Anybody who has tried to parse CEO compensation in recent years knows that more disclosure doesn't guarantee clarity, but any increase in information in the CDS realm will be a benefit. Perhaps that would limit the baleful effect of CDS on (must we consider it?) the next disaster - or even help us prevent it.
mfg ipollit
http://money.cnn.com/2008/09/30/magazines/fortune/varchaver_…
The $55 trillion question
The financial crisis has put a spotlight on the obscure world of credit default swaps - which trade in a vast, unregulated market that most people haven't heard of and even fewer understand. Will this be the next disaster?
By Nicholas Varchaver, senior editor and Katie Benner, writer-reporter
Last Updated: September 30, 2008: 12:28 PM ET
(Fortune Magazine) -- As Congress wrestles with another bailout bill to try to contain the financial contagion, there's a potential killer bug out there whose next movement can't be predicted: the Credit Default Swap.
In just over a decade these privately traded derivatives contracts have ballooned from nothing into a $54.6 trillion market. CDS are the fastest-growing major type of financial derivatives. More important, they've played a critical role in the unfolding financial crisis. First, by ostensibly providing "insurance" on risky mortgage bonds, they encouraged and enabled reckless behavior during the housing bubble.
"If CDS had been taken out of play, companies would've said, 'I can't get this [risk] off my books,'" says Michael Greenberger, a University of Maryland law professor and former director of trading and markets at the Commodity Futures Trading Commission. "If they couldn't keep passing the risk down the line, those guys would've been stopped in their tracks. The ultimate assurance for issuing all this stuff was, 'It's insured.'"
Second, terror at the potential for a financial Ebola virus radiating out from a failing institution and infecting dozens or hundreds of other companies - all linked to one another by CDS and other instruments - was a major reason that regulators stepped in to bail out Bear Stearns and buy out AIG (AIG, Fortune 500), whose calamitous descent itself was triggered by losses on its CDS contracts (see "Hank's Last Stand").
And the fear of a CDS catastrophe still haunts the markets. For starters, nobody knows how federal intervention might ripple through this chain of contracts. And meanwhile, as we'll see, two fundamental aspects of the CDS market - that it is unregulated, and that almost nothing is disclosed publicly - may be about to change. That adds even more uncertainty to the equation.
"The big problem is that here are all these public companies - banks and corporations - and no one really knows what exposure they've got from the CDS contracts," says Frank Partnoy, a law professor at the University of San Diego and former Morgan Stanley derivatives salesman who has been writing about the dangers of CDS and their ilk for a decade. "The really scary part is that we don't have a clue." Chris Wolf, a co-manager of Cogo Wolf, a hedge fund of funds, compares them to one of the great mysteries of astrophysics: "This has become essentially the dark matter of the financial universe."
***
AT FIRST GLANCE, credit default swaps don't look all that scary. A CDS is just a contract: The "buyer" plunks down something that resembles a premium, and the "seller" agrees to make a specific payment if a particular event, such as a bond default, occurs. Used soberly, CDS offer concrete benefits: If you're holding bonds and you're worried that the issuer won't be able to pay, buying CDS should cover your loss. "CDS serve a very useful function of allowing financial markets to efficiently transfer credit risk," argues Sunil Hirani, the CEO of Creditex, one of a handful of marketplaces that trade the contracts.
Because they're contracts rather than securities or insurance, CDS are easy to create: Often deals are done in a one-minute phone conversation or an instant message. Many technical aspects of CDS, such as the typical five-year term, have been standardized by the International Swaps and Derivatives Association (ISDA). That only accelerates the process. You strike your deal, fill out some forms, and you've got yourself a $5 million - or a $100 million - contract.
And as long as someone is willing to take the other side of the proposition, a CDS can cover just about anything, making it the Wall Street equivalent of those notorious Lloyds of London policies covering Liberace's hands and other esoterica. It has even become possible to purchase a CDS that would pay out if the U.S. government defaults. (Trust us when we say that if the government goes under, trying to collect will be the least of your worries.)
You can guess how Wall Street cowboys responded to the opportunity to make deals that (1) can be struck in a minute, (2) require little or no cash upfront, and (3) can cover anything. Yee-haw! You can almost picture Slim Pickens in Dr. Strangelove climbing onto the H-bomb before it's released from the B-52. And indeed, the volume of CDS has exploded with nuclear force, nearly doubling every year since 2001 to reach a recent peak of $62 trillion at the end of 2007, before receding to $54.6 trillion as of June 30, according to ISDA.
Take that gargantuan number with a grain of salt. It refers to the face value of all outstanding contracts. But many players in the market hold offsetting positions. So if, in theory, every entity that owns CDS had to settle its contracts tomorrow and "netted" all its positions against each other, a much smaller amount of money would change hands. But even a tiny fraction of that $54.6 trillion would still be a daunting sum.
The credit freeze and then the Bear disaster explain the drop in outstanding CDS contracts during the first half of the year - and the market has only worsened since. CDS contracts on widely held debt, such as General Motors' (GM, Fortune 500), continue to be actively bought and sold. But traders say almost no new contracts are being written on any but the most liquid debt issues right now, in part because nobody wants to put money at risk and because nobody knows what Washington will do and how that will affect the market. ("There's nothing to do but watch Bernanke on TV," one trader told Fortune during the week when the Fed chairman was going before Congress to push the mortgage bailout.) So, after nearly a decade of exponential growth, the CDS market is poised for its first sustained contraction.
***
ONE REASON THE MARKET TOOK OFF is that you don't have to own a bond to buy a CDS on it - anyone can place a bet on whether a bond will fail. Indeed the majority of CDS now consists of bets on other people's debt. That's why it's possible for the market to be so big: The $54.6 trillion in CDS contracts completely dwarfs total corporate debt, which the Securities Industry and Financial Markets Association puts at $6.2 trillion, and the $10 trillion it counts in all forms of asset-backed debt.
"It's sort of like I think you're a bad driver and you're going to crash your car," says Greenberger, formerly of the CFTC. "So I go to an insurance company and get collision insurance on your car because I think it'll crash and I'll collect on it." That's precisely what the biggest winners in the subprime debacle did. Hedge fund star John Paulson of Paulson & Co., for example, made $15 billion in 2007, largely by using CDS to bet that other investors' subprime mortgage bonds would default.
So what started out as a vehicle for hedging ended up giving investors a cheap, easy way to wager on almost any event in the credit markets. In effect, credit default swaps became the world's largest casino. As Christopher Whalen, a managing director of Institutional Risk Analytics, observes, "To be generous, you could call it an unregulated, uncapitalized insurance market. But really, you would call it a gaming contract."
There is at least one key difference between casino gambling and CDS trading: Gambling has strict government regulation. The federal government has long shied away from any oversight of CDS. The CFTC floated the idea of taking an oversight role in the late '90s, only to find itself opposed by Federal Reserve chairman Alan Greenspan and others. Then, in 2000, Congress, with the support of Greenspan and Treasury Secretary Lawrence Summers, passed a bill prohibiting all federal and most state regulation of CDS and other derivatives. In a press release at the time, co-sponsor Senator Phil Gramm - most recently in the news when he stepped down as John McCain's campaign co-chair this summer after calling people who talk about a recession "whiners" - crowed that the new law "protects financial institutions from over-regulation ... and it guarantees that the United States will maintain its global dominance of financial markets." (The authors of the legislation were so bent on warding off regulation that they had the bill specify that it would "supersede and preempt the application of any state or local law that prohibits gaming ...") Not everyone was as sanguine as Gramm. In 2003 Warren Buffett famously called derivatives "financial weapons of mass destruction."
***
THERE'S ANOTHER BIG difference between trading CDS and casino gambling. When you put $10 on black 22, you're pretty sure the casino will pay off if you win. The CDS market offers no such assurance. One reason the market grew so quickly was that hedge funds poured in, sensing easy money. And not just big, well-established hedge funds but a lot of upstarts. So in some cases, giant financial institutions were counting on collecting money from institutions only slightly more solvent than your average minimart. The danger, of course, is that if a hedge fund suddenly has to pay off on a lot of CDS, it will simply go out of business. "People have been insuring risks that they can't insure," says Peter Schiff, the president of Euro Pacific Capital and author of Crash Proof, which predicted doom for Fannie and Freddie, among other things. "Let's say you're writing fire insurance policies, and every time you get the [premium], you spend it. You just assume that no houses are going to burn down. And all of a sudden there's a huge fire and they all burn down. What do you do? You just close up shop."
This is not an academic concern. Wachovia (WB, Fortune 500) and Citigroup (C, Fortune 500) are wrangling in court with a $50 million hedge fund located in the Channel Islands. The reason: A dispute over two $10 million credit default swaps covering some CDOs. The specifics of the spat aren't important. What's most revealing is that these massive banks put their faith in a Lilliputian fund (in an inaccessible jurisdiction) that was risking 40% of its capital for just two CDS. Can anyone imagine that Citi would, say, insure its headquarters building with a thinly capitalized, unregulated, offshore entity?
That's one element of what's known as "counterparty risk." Here's another: In many cases, you don't even know who has the other side of your bet. Parties to the contract can, and do, transfer their side of the contract to third parties. Investment firms assert that transfers are well documented (a claim that, like most in the world of CDS, is impossible to verify). But even if that's true, you're still left with the fact that a given company's risks are being dispersed in ways that they may not know about and can't control.
It doesn't help that CDS trading is a haphazard process. Most contracts are bought and sold over the phone or by instant message and settled manually. Settlement has been sloppy, confirms Jamie Cawley of IDX Capital, a firm that brokers trades between big banks. Pushed by New York Fed president Timothy Geithner, the players have been improving the process. But even as recently as a year ago, Cawley says, so many trades were sitting around unfulfilled that "there were $1 trillion worth of swaps that were unsettled among counterparties."
Trade settlement is not the only anachronistic aspect of CDS trading. Consider what will happen with CDS contracts relating to Fannie Mae and Freddie Mac. The two were placed in conservatorship on Sept. 7. But the value of many contracts won't be determined till Oct. 6, when an auction will set a cash price for Fannie and Freddie bonds. We'll spare you the technical reasons, but suffice it to ask: Can you imagine any other major market that would need a month to resolve something like this?
***
WITH WASHINGTON SUDDENLY in a frenzy of outrage over the financial markets, debating everything from the shape and extent of the mortgage plan to what should be done about short-selling, the future for CDS is very blurry. "The market is here to stay," asserts Cawley. The question is simply: What sorts of changes are in store? As this article was going to press, SEC chairman Christopher Cox asked the Senate to allow his agency to begin regulating CDS - mostly, it should be said, to rein in short-selling. And the SEC separately announced that it was expanding its investigation of market manipulation, which initially targeted the short-sellers, to CDS investors.
Under other circumstances, Cox's request might have been met with polite silence. But the convulsions over the mortgage bailout are so dramatic that they are reminiscent of the moment, soon after the Enron scandal, when Congress drafted the Sarbanes-Oxley legislation. The desire to blame short-sellers may actually result in powers for Cox that, until very recently, he showed no signs of wanting. Should legislators wade into this issue, the measures most widely seen as necessary are straightforward: some form of centralized trading or clearing and some form of capital or reserve requirements. Meanwhile, New York State's insurance commissioner, Eric Dinallo, announced new regulations that would essentially treat sellers of some (but not all) CDS as insurance entities, thereby forcing them to set aside reserves and otherwise follow state insurance law - requirements that would probably drive many participants from the market. Whether CDS players will find a way to challenge the rules remains to be seen. (ISDA, the industry's trade group, has already gone on record in opposition to Cox's proposal.) If nothing else, the New York law may provide additional impetus for the feds to take action.
For now, the biggest impact could come from the Financial Accounting Standards Board. It is implementing a new rule in November that will require sellers of CDS and other credit derivatives to report detailed information, including their maximum payouts and reasons for entering the contracts, as well as assets that might allow them to offset any payouts. Anybody who has tried to parse CEO compensation in recent years knows that more disclosure doesn't guarantee clarity, but any increase in information in the CDS realm will be a benefit. Perhaps that would limit the baleful effect of CDS on (must we consider it?) the next disaster - or even help us prevent it.
mfg ipollit
Antwort auf Beitrag Nr.: 35.395.665 von ipollit am 03.10.08 13:25:18http://www.handelsblatt.com/finanzen/finanzmarktprofis/die-a…
26.09.2008
Rieckes Rückfrage
Die AIG-Connection
von Torsten Riecke
Die Reaktion von Bundesfinanzminister Peer Steinbrück auf die Bitte der USA, sich an dem Rettungsplan für das globale Finanzsystem zu beteiligen, ist eindeutig: you broke it, you own it. Auf gut Deutsch: die Suppe müsst ihr alleine auslöffeln. Aus anderen europäischen Hauptstädten kommen ähnliche Signale.
Damit machen wir Europäer uns die Sache jedoch wieder einmal zu einfach. Nehmen wir das Beispiel AIG. Der beinahe pleitegegangene US-Versicherungskonzern hatte Ende Juni ein Volumen von etwa 440 Mrd. Dollar sogenannter "Credit Default Swaps" (CDS) in seinen Büchern. Bei diesen obskuren Finanzprodukten handelt es sich um eine Art Kreditversicherung für Anleihen, die dem Käufer bei einem Zahlungsausfall zur Seite springt.
Der verständliche Wunsch nach einer Risikovorsorge von Gläubigern diente findigen Finanzakrobaten jedoch als Vehikel, ein internationales Spielkasino zu eröffnen. Die CDS wurden für Bonitätswetten auf Anleiheprodukte aller Art missbraucht, um damit kurzfristige Profite einzufahren. So ist der Markt für diese Kasinochips auf heute 62 Bill. Dollar gewachsen. Das bunte Treiben findet nach Worten der US-Börsenaufsicht SEC ohne jegliche Aufsicht statt.
Wer sind die Spieler in diesem Finanzroulette? AIG, soviel wissen wir bereits, gehörte zu den risikofreudigsten Firmen. Was nach außen den Anschein einer soliden Versicherung mit spröden Hausrat- und Autopolicen hatte, war in Wirklichkeit ein riesiger, unkontrollierter Hedge-Fonds. Doch wer waren die Mitspieler? Nach Angaben von AIG wurden etwa zwei Drittel der von ihr ausgestellten CDS von europäischen Banken aufgekauft, um sich gegen Zahlungsausfälle von Anleihen aus dem US-Hypothekenmarkt abzusichern. Über die Hintergründe dieser obskuren Absicherung wird heftig gestritten. Viele Experten glauben, die europäischen Finanzinstitute hätten sich nur deshalb der CDS bedient, um die Kapitalanforderungen nach den Basel-II-Vorschriften zu umgehen. Der Fachmann spricht von "regulatory arbitrage", man kann das aber auch als Taschenspielertrick geißeln. Fakt ist, dass die Rettung von AIG auch die europäischen Banken vor dem Schlimmsten bewahrt hat. Kann Europa es sich angesichts der AIG-Connection wirklich leisten, bei der Rettung des Finanzsystems abseits zu stehen?
************
http://www.handelsblatt.com/finanzen/breakingviews/trennen-w…
26.09.2008
Kreditderivate
Trennen, was verbindet
von Richard Beales, breakingviews.com
Der Markt für Credit Default Swaps (CDS) ist in den ersten sechs Monaten dieses Jahres tatsächlich geschrumpft - auf "nur" 55 Bill. Dollar. Das ist ein Grund zum Aufatmen, denn diese Kreditderivate zum Handeln von Ausfallrisiken von Krediten und Anleihen befinden sich im Fadenkreuz der Aufsichtsbehörden und waren teilweise für die finanziellen Schwierigkeiten von Bear Stearns und American International Group (AIG) verantwortlich gemacht worden. Doch während dies als ein gutes Zeichen gewertet werden mag, dass unnötiger Ärger vom Markt ferngehalten wird, werden immer noch mehr und mehr Risiken eingegangen. Und die Marktregulierer werden wahrscheinlich so schnell nicht nachgeben.
Die genannte Zahl spiegelt das gesamte Kreditengagement sämtlicher global umlaufender CDS-Kontrakte wider. Sie ist gegenüber dem Betrag von 62 Bill. Dollar, der Ende 2007 erreicht worden war, um zwölf Prozent gefallen, wie Umfragen der International Swaps and Derivatives Association zu entnehmen ist. Das ist gut - bedeutet dies doch, dass da draußen weniger CDS-Kontrakte herumgereicht werden, wodurch weniger logistische Irrtümer und rechtliche Verstrickungen zu erwarten sind.
Sie zeigt allerdings nicht, dass sich der Handel mit diesen Papieren verlangsamt hätte. Berechnungen von Markit, einer Gruppe, die Daten zu den Derivat-Märkten auswertet, zeigen, dass die monatlichen CDS-Aktivitäten im ersten Halbjahr immer noch Aufwärtstendenzen aufwiesen, wenn auch keine Rekordhochs mehr erreicht wurden. Aber die Branche hat sich auch bei dem Versuch ganz besonders ins Zeug gelegt, ausgleichende Positionen zu neutralisieren, die sonst gesondert erfasst worden wären. Trioptima, eine Firma, die bei diesem Prozess behilflich ist, hat im Berichtszeitraum "Aufkündigungen" dieser Art über 17 Bill. Dollar vorgenommen.
Diese Positionen wieder hinzuzuaddieren, würde wahrscheinlich einen realistischeren Vergleich mit den Zahlen vom Ende des vergangenen Jahres liefern. Unter dieser Voraussetzung wäre das nominelle ausstehende CDS-Engagement zwischen Dezember und Juni um 16 Prozent auf 72 Bill. Dollar gestiegen.
Damit wird das tatsächlich eingegangene Risiko enorm übertrieben dargestellt. Es ist keine exakte Wissenschaft, aber die Bank für Internationalen Zahlungsausgleich geht davon aus, dass der Marktwert der ausstehenden CDS-Kontrakte im Dezember 3,5 Prozent des zugrunde liegenden Engagements betragen hatte. Wendet man dies auf die Zahl von 72 Bill. Dollar an, dann könnte der Marktwert der CDS-Kontrakte per Ende Juni bei etwa 2,5 Bill. Dollar gelegen haben.
Das hört sich schon nicht mehr so beängstigend an, aber es ist immer noch viel Geld, das gewonnen und verloren werden kann - zusammen mit der zusätzlichen Sorge darüber, dass ins Straucheln geratene CDS-Handelspartner eventuell ihre Kontrakte nicht ausgleichen. Daher, und besonders nach den Vorfällen bei Bear Stearns und AIG, dürfte der private CDS-Markt wahrscheinlich einer genaueren Beobachtung durch die Aufsichtsbehörden nicht entkommen.
Jüngste Wortmeldungen seitens der Aufsichtsbehörden für die Versicherungsbranche des US-Bundesstaats New York und der US-Wertpapieraufsicht Securities and Exchange Commission mögen zu wenig oder zu gar nichts führen. Aber wenigstens könnten sie die CDS-Industrie dazu ermutigen, die praktische Lösung, die die Federal Reserve von New York bevorzugt, schnell voranzutreiben, um den Markt zu straffen und systemische Gefahren zu reduzieren: Sie sollten eine zentrale Abrechnungsstelle einrichten.
mfg ipollit
26.09.2008
Rieckes Rückfrage
Die AIG-Connection
von Torsten Riecke
Die Reaktion von Bundesfinanzminister Peer Steinbrück auf die Bitte der USA, sich an dem Rettungsplan für das globale Finanzsystem zu beteiligen, ist eindeutig: you broke it, you own it. Auf gut Deutsch: die Suppe müsst ihr alleine auslöffeln. Aus anderen europäischen Hauptstädten kommen ähnliche Signale.
Damit machen wir Europäer uns die Sache jedoch wieder einmal zu einfach. Nehmen wir das Beispiel AIG. Der beinahe pleitegegangene US-Versicherungskonzern hatte Ende Juni ein Volumen von etwa 440 Mrd. Dollar sogenannter "Credit Default Swaps" (CDS) in seinen Büchern. Bei diesen obskuren Finanzprodukten handelt es sich um eine Art Kreditversicherung für Anleihen, die dem Käufer bei einem Zahlungsausfall zur Seite springt.
Der verständliche Wunsch nach einer Risikovorsorge von Gläubigern diente findigen Finanzakrobaten jedoch als Vehikel, ein internationales Spielkasino zu eröffnen. Die CDS wurden für Bonitätswetten auf Anleiheprodukte aller Art missbraucht, um damit kurzfristige Profite einzufahren. So ist der Markt für diese Kasinochips auf heute 62 Bill. Dollar gewachsen. Das bunte Treiben findet nach Worten der US-Börsenaufsicht SEC ohne jegliche Aufsicht statt.
Wer sind die Spieler in diesem Finanzroulette? AIG, soviel wissen wir bereits, gehörte zu den risikofreudigsten Firmen. Was nach außen den Anschein einer soliden Versicherung mit spröden Hausrat- und Autopolicen hatte, war in Wirklichkeit ein riesiger, unkontrollierter Hedge-Fonds. Doch wer waren die Mitspieler? Nach Angaben von AIG wurden etwa zwei Drittel der von ihr ausgestellten CDS von europäischen Banken aufgekauft, um sich gegen Zahlungsausfälle von Anleihen aus dem US-Hypothekenmarkt abzusichern. Über die Hintergründe dieser obskuren Absicherung wird heftig gestritten. Viele Experten glauben, die europäischen Finanzinstitute hätten sich nur deshalb der CDS bedient, um die Kapitalanforderungen nach den Basel-II-Vorschriften zu umgehen. Der Fachmann spricht von "regulatory arbitrage", man kann das aber auch als Taschenspielertrick geißeln. Fakt ist, dass die Rettung von AIG auch die europäischen Banken vor dem Schlimmsten bewahrt hat. Kann Europa es sich angesichts der AIG-Connection wirklich leisten, bei der Rettung des Finanzsystems abseits zu stehen?
************
http://www.handelsblatt.com/finanzen/breakingviews/trennen-w…
26.09.2008
Kreditderivate
Trennen, was verbindet
von Richard Beales, breakingviews.com
Der Markt für Credit Default Swaps (CDS) ist in den ersten sechs Monaten dieses Jahres tatsächlich geschrumpft - auf "nur" 55 Bill. Dollar. Das ist ein Grund zum Aufatmen, denn diese Kreditderivate zum Handeln von Ausfallrisiken von Krediten und Anleihen befinden sich im Fadenkreuz der Aufsichtsbehörden und waren teilweise für die finanziellen Schwierigkeiten von Bear Stearns und American International Group (AIG) verantwortlich gemacht worden. Doch während dies als ein gutes Zeichen gewertet werden mag, dass unnötiger Ärger vom Markt ferngehalten wird, werden immer noch mehr und mehr Risiken eingegangen. Und die Marktregulierer werden wahrscheinlich so schnell nicht nachgeben.
Die genannte Zahl spiegelt das gesamte Kreditengagement sämtlicher global umlaufender CDS-Kontrakte wider. Sie ist gegenüber dem Betrag von 62 Bill. Dollar, der Ende 2007 erreicht worden war, um zwölf Prozent gefallen, wie Umfragen der International Swaps and Derivatives Association zu entnehmen ist. Das ist gut - bedeutet dies doch, dass da draußen weniger CDS-Kontrakte herumgereicht werden, wodurch weniger logistische Irrtümer und rechtliche Verstrickungen zu erwarten sind.
Sie zeigt allerdings nicht, dass sich der Handel mit diesen Papieren verlangsamt hätte. Berechnungen von Markit, einer Gruppe, die Daten zu den Derivat-Märkten auswertet, zeigen, dass die monatlichen CDS-Aktivitäten im ersten Halbjahr immer noch Aufwärtstendenzen aufwiesen, wenn auch keine Rekordhochs mehr erreicht wurden. Aber die Branche hat sich auch bei dem Versuch ganz besonders ins Zeug gelegt, ausgleichende Positionen zu neutralisieren, die sonst gesondert erfasst worden wären. Trioptima, eine Firma, die bei diesem Prozess behilflich ist, hat im Berichtszeitraum "Aufkündigungen" dieser Art über 17 Bill. Dollar vorgenommen.
Diese Positionen wieder hinzuzuaddieren, würde wahrscheinlich einen realistischeren Vergleich mit den Zahlen vom Ende des vergangenen Jahres liefern. Unter dieser Voraussetzung wäre das nominelle ausstehende CDS-Engagement zwischen Dezember und Juni um 16 Prozent auf 72 Bill. Dollar gestiegen.
Damit wird das tatsächlich eingegangene Risiko enorm übertrieben dargestellt. Es ist keine exakte Wissenschaft, aber die Bank für Internationalen Zahlungsausgleich geht davon aus, dass der Marktwert der ausstehenden CDS-Kontrakte im Dezember 3,5 Prozent des zugrunde liegenden Engagements betragen hatte. Wendet man dies auf die Zahl von 72 Bill. Dollar an, dann könnte der Marktwert der CDS-Kontrakte per Ende Juni bei etwa 2,5 Bill. Dollar gelegen haben.
Das hört sich schon nicht mehr so beängstigend an, aber es ist immer noch viel Geld, das gewonnen und verloren werden kann - zusammen mit der zusätzlichen Sorge darüber, dass ins Straucheln geratene CDS-Handelspartner eventuell ihre Kontrakte nicht ausgleichen. Daher, und besonders nach den Vorfällen bei Bear Stearns und AIG, dürfte der private CDS-Markt wahrscheinlich einer genaueren Beobachtung durch die Aufsichtsbehörden nicht entkommen.
Jüngste Wortmeldungen seitens der Aufsichtsbehörden für die Versicherungsbranche des US-Bundesstaats New York und der US-Wertpapieraufsicht Securities and Exchange Commission mögen zu wenig oder zu gar nichts führen. Aber wenigstens könnten sie die CDS-Industrie dazu ermutigen, die praktische Lösung, die die Federal Reserve von New York bevorzugt, schnell voranzutreiben, um den Markt zu straffen und systemische Gefahren zu reduzieren: Sie sollten eine zentrale Abrechnungsstelle einrichten.
mfg ipollit
Antwort auf Beitrag Nr.: 35.395.753 von ipollit am 03.10.08 13:30:01http://www.moneymorning.com/2008/09/18/credit-default-swaps/
Thursday, September 18th, 2008
The Real Reason for the Global Financial Crisis…the Story No One’s Talking About
[Part I of a three-part series looking at how so-called “credit default swap” derivatives could ignite a worldwide capital markets meltdown.]
By Shah Gilani
Contributing Editor
Are you shell-shocked? Are you wondering what’s really going on in the market? The truth is probably more frightening than even your worst fears. And yet, you won’t hear about it anywhere else because “they” can’t tell you. “They” are the U.S. Federal Reserve and the U.S. Treasury Department, and they can’t tell you what’s really going on because there’s nothing they can do about it, except what they’ve been trying to do - add liquidity.
At the exchange rate yesterday (Wednesday), 35 trillion British Pounds was equivalent to U.S. $62 trillion (hence, the 35 trillion Pound gorilla). According to the International Swaps and Derivatives Association, $62 trillion is the notional value of credit default swaps (CDS) out there, somewhere, in the market.
This isn’t the first time Money Morning has warned readers about the dangers of credit default swaps. And it won’t be the last.
The Genesis of a Derivative Boom
In the mid-1980s, upon arriving in New York from Chicago with an extensive background trading options and futures (the original derivatives), I was offered a job at what was then Citicorp [today's Citigroup Inc. (C)]. The offer was for an entry-level post in the bank’s brand new OTC (over-the-counter, meaning not exchange traded) swaps and derivatives group. When I asked what the economic purpose of swaps was, the answer came back: “To make money for the bank.”
I declined the position.
It used to be that regulators and legislators demanded theoretical, empirical, and quantitative measures of the efficacy of new tradable instruments being proposed by exchanges. What is their purpose? How will they benefit the capital markets and the economy? And, what safeguards will accompany their introduction?
Not any more. In the early 1990s, in order to hedge their loan risks, J. P. Morgan & Co. [now JPMorgan Chase & Co. (JPM)] bankers devised credit default swaps.
A credit default swap is, essentially, an insurance contract between a protection buyer and a protection seller covering a corporation’s, or sovereign’s (the “referenced entity”), specific bond or loan. A protection buyer pays an upfront amount and yearly premiums to the protection seller to cover any loss on the face amount of the referenced bond or loan.
Typically, the insurance is for five years.
Credit default swaps are bilateral contracts, meaning they are private contracts between two parties. CDSs are subject only to the collateral and margin agreed to by contract. They are traded over-the-counter, usually by telephone. They are subject to re-sale to another party willing to enter into another contract. Most frighteningly, credit default swaps are subject to “counterparty risk.”
If the party providing the insurance protection - once it has collected its upfront payment and premiums - doesn’t have the money to pay the insured buyer in the case of a default event affecting the referenced bond or loan (think hedge funds), or if the “insurer” goes bankrupt (Bear Stearns was almost there, and American International Group Inc. (AIG) was almost there) the buyer is not covered - period. The premium payments are gone, as is the insurance against default.
Credit default swaps are not standardized instruments. In fact, they technically aren’t true securities in the classic sense of the word in that they’re not transparent, aren’t traded on any exchange, aren’t subject to present securities laws, and aren’t regulated. They are, however, at risk - all $62 trillion (the best guess by the ISDA) of them.
Fundamentally, this kind of derivative serves a real purpose - as a hedging device. The actual holders, or creditors, of outstanding corporate or sovereign loans and bonds might seek insurance to guarantee that the debts they are owed are repaid. That’s the economic purpose of insurance.
What happened, however, is that risk speculators who wanted exposure to certain asset classes, various bonds and loans, or security pools such as residential and commercial mortgage-backed securities (yes, those same subprime mortgage-backed securities that you’ve been reading about), but didn’t actually own the underlying credits, now had a means by which to speculate on them.
If you think XYZ Corp. is in trouble, and won’t be able to pay back its bondholders, you can speculate by buying, and paying premiums for, credit default swaps on their bonds, which will pay you the full face amount of the bonds if they do actually default. If, on the other hand, you think that XYZ Corp. is doing just fine, and its bonds are as good as gold, you can offer insurance to a fellow speculator, who holds the opinion opposite yours. That means you’d essentially be speculating that the bonds would not default. You’re hoping that you’ll collect, and keep, all the premiums, and never have to pay off on the insurance. It’s pure speculation.
Credit default swaps are not unlike me being able to insure your house, not with you, but with someone else entirely not connected to your house, so that if your house is washed away in the next hurricane I get paid its value. I’m speculating on an event. I’m making a bet.
The bad news is that there are even worse bets out there. There are credit default swaps written on subprime mortgage securities. It’s bad enough that these subprime mortgage pools that banks, investment banks, insurance companies, hedge funds and others bought were over-rated and ended up falling precipitously in value as foreclosures mounted on the underlying mortgages in the pools.
What’s even worse, however, is that speculators sold and bought trillions of dollars of insurance that these pools would, or wouldn’t, default! The sellers of this insurance (AIG is one example) are getting killed as defaults continue to rise with no end in sight.
And this is only where the story begins.
The Ticking Time Bomb
What is happening in both the stock and credit markets is a direct result of what’s playing out in the CDS market. The Fed could not let Bear Stearns enter bankruptcy because - and only because - the trillions of dollars of credit default swaps on its books would be wiped out. All the banks and institutions that had insurance written by Bear would not be able to say that they were insured or hedged anymore and they would have to write-down billions and billions of dollars in losses that they’ve been carrying at higher values because they could say that they were insured for those losses.
The counterparty risk that all Bear’s trading partners were exposed to was so far and wide, and so deep, that if Bear was to enter bankruptcy it would take years to sort out the risk and losses. That was an untenable option.
The Fed had to bail out Bear Stearns.
The same thing has just happened to AIG. Make no mistake about it, there’s nothing wrong with AIG’s insurance subsidiaries - absolutely nothing. In fact, the Fed just made the best trade in its history by bailing AIG out and getting equity, warrants and charging the insurance giant seven points over the benchmark London Interbank Offered Rate (LIBOR) on that $85 billion loan!
What happened to AIG is simple: AIG got greedy. AIG, as of June 30, had written $441 billion worth of swaps on corporate bonds, and worse, mortgage-backed securities. As the value of these insured-referenced entities fell, AIG had massive write-downs and additionally had to post more collateral. And when its ratings were downgraded on Monday evening, the company had to post even more collateral, which it didn’t have.
In short, what happened in one small AIG corporate subsidiary blew apart the largest insurance company in the world.
But there’s more - a lot more. These instruments are causing many of the massive write-downs at banks, investment banks and insurance companies. Knowing what all this means for hedge funds, the credit markets and the stock market is the key to understanding where this might end and how.
The rest of the story will be illuminated in the next two installments. Next up: An examination of the AIG collapse, followed by a look at how bad things could get, and what we can do to fix the problem at hand. So stay tuned.
mfg ipollit
Thursday, September 18th, 2008
The Real Reason for the Global Financial Crisis…the Story No One’s Talking About
[Part I of a three-part series looking at how so-called “credit default swap” derivatives could ignite a worldwide capital markets meltdown.]
By Shah Gilani
Contributing Editor
Are you shell-shocked? Are you wondering what’s really going on in the market? The truth is probably more frightening than even your worst fears. And yet, you won’t hear about it anywhere else because “they” can’t tell you. “They” are the U.S. Federal Reserve and the U.S. Treasury Department, and they can’t tell you what’s really going on because there’s nothing they can do about it, except what they’ve been trying to do - add liquidity.
At the exchange rate yesterday (Wednesday), 35 trillion British Pounds was equivalent to U.S. $62 trillion (hence, the 35 trillion Pound gorilla). According to the International Swaps and Derivatives Association, $62 trillion is the notional value of credit default swaps (CDS) out there, somewhere, in the market.
This isn’t the first time Money Morning has warned readers about the dangers of credit default swaps. And it won’t be the last.
The Genesis of a Derivative Boom
In the mid-1980s, upon arriving in New York from Chicago with an extensive background trading options and futures (the original derivatives), I was offered a job at what was then Citicorp [today's Citigroup Inc. (C)]. The offer was for an entry-level post in the bank’s brand new OTC (over-the-counter, meaning not exchange traded) swaps and derivatives group. When I asked what the economic purpose of swaps was, the answer came back: “To make money for the bank.”
I declined the position.
It used to be that regulators and legislators demanded theoretical, empirical, and quantitative measures of the efficacy of new tradable instruments being proposed by exchanges. What is their purpose? How will they benefit the capital markets and the economy? And, what safeguards will accompany their introduction?
Not any more. In the early 1990s, in order to hedge their loan risks, J. P. Morgan & Co. [now JPMorgan Chase & Co. (JPM)] bankers devised credit default swaps.
A credit default swap is, essentially, an insurance contract between a protection buyer and a protection seller covering a corporation’s, or sovereign’s (the “referenced entity”), specific bond or loan. A protection buyer pays an upfront amount and yearly premiums to the protection seller to cover any loss on the face amount of the referenced bond or loan.
Typically, the insurance is for five years.
Credit default swaps are bilateral contracts, meaning they are private contracts between two parties. CDSs are subject only to the collateral and margin agreed to by contract. They are traded over-the-counter, usually by telephone. They are subject to re-sale to another party willing to enter into another contract. Most frighteningly, credit default swaps are subject to “counterparty risk.”
If the party providing the insurance protection - once it has collected its upfront payment and premiums - doesn’t have the money to pay the insured buyer in the case of a default event affecting the referenced bond or loan (think hedge funds), or if the “insurer” goes bankrupt (Bear Stearns was almost there, and American International Group Inc. (AIG) was almost there) the buyer is not covered - period. The premium payments are gone, as is the insurance against default.
Credit default swaps are not standardized instruments. In fact, they technically aren’t true securities in the classic sense of the word in that they’re not transparent, aren’t traded on any exchange, aren’t subject to present securities laws, and aren’t regulated. They are, however, at risk - all $62 trillion (the best guess by the ISDA) of them.
Fundamentally, this kind of derivative serves a real purpose - as a hedging device. The actual holders, or creditors, of outstanding corporate or sovereign loans and bonds might seek insurance to guarantee that the debts they are owed are repaid. That’s the economic purpose of insurance.
What happened, however, is that risk speculators who wanted exposure to certain asset classes, various bonds and loans, or security pools such as residential and commercial mortgage-backed securities (yes, those same subprime mortgage-backed securities that you’ve been reading about), but didn’t actually own the underlying credits, now had a means by which to speculate on them.
If you think XYZ Corp. is in trouble, and won’t be able to pay back its bondholders, you can speculate by buying, and paying premiums for, credit default swaps on their bonds, which will pay you the full face amount of the bonds if they do actually default. If, on the other hand, you think that XYZ Corp. is doing just fine, and its bonds are as good as gold, you can offer insurance to a fellow speculator, who holds the opinion opposite yours. That means you’d essentially be speculating that the bonds would not default. You’re hoping that you’ll collect, and keep, all the premiums, and never have to pay off on the insurance. It’s pure speculation.
Credit default swaps are not unlike me being able to insure your house, not with you, but with someone else entirely not connected to your house, so that if your house is washed away in the next hurricane I get paid its value. I’m speculating on an event. I’m making a bet.
The bad news is that there are even worse bets out there. There are credit default swaps written on subprime mortgage securities. It’s bad enough that these subprime mortgage pools that banks, investment banks, insurance companies, hedge funds and others bought were over-rated and ended up falling precipitously in value as foreclosures mounted on the underlying mortgages in the pools.
What’s even worse, however, is that speculators sold and bought trillions of dollars of insurance that these pools would, or wouldn’t, default! The sellers of this insurance (AIG is one example) are getting killed as defaults continue to rise with no end in sight.
And this is only where the story begins.
The Ticking Time Bomb
What is happening in both the stock and credit markets is a direct result of what’s playing out in the CDS market. The Fed could not let Bear Stearns enter bankruptcy because - and only because - the trillions of dollars of credit default swaps on its books would be wiped out. All the banks and institutions that had insurance written by Bear would not be able to say that they were insured or hedged anymore and they would have to write-down billions and billions of dollars in losses that they’ve been carrying at higher values because they could say that they were insured for those losses.
The counterparty risk that all Bear’s trading partners were exposed to was so far and wide, and so deep, that if Bear was to enter bankruptcy it would take years to sort out the risk and losses. That was an untenable option.
The Fed had to bail out Bear Stearns.
The same thing has just happened to AIG. Make no mistake about it, there’s nothing wrong with AIG’s insurance subsidiaries - absolutely nothing. In fact, the Fed just made the best trade in its history by bailing AIG out and getting equity, warrants and charging the insurance giant seven points over the benchmark London Interbank Offered Rate (LIBOR) on that $85 billion loan!
What happened to AIG is simple: AIG got greedy. AIG, as of June 30, had written $441 billion worth of swaps on corporate bonds, and worse, mortgage-backed securities. As the value of these insured-referenced entities fell, AIG had massive write-downs and additionally had to post more collateral. And when its ratings were downgraded on Monday evening, the company had to post even more collateral, which it didn’t have.
In short, what happened in one small AIG corporate subsidiary blew apart the largest insurance company in the world.
But there’s more - a lot more. These instruments are causing many of the massive write-downs at banks, investment banks and insurance companies. Knowing what all this means for hedge funds, the credit markets and the stock market is the key to understanding where this might end and how.
The rest of the story will be illuminated in the next two installments. Next up: An examination of the AIG collapse, followed by a look at how bad things could get, and what we can do to fix the problem at hand. So stay tuned.
mfg ipollit
Antwort auf Beitrag Nr.: 35.395.834 von ipollit am 03.10.08 13:34:06http://www.moneymorning.com/2008/09/22/credit-default-swaps-…
Monday, September 22nd, 2008
The Credit Crisis and the Real Story Behind the Collapse of AIG
[In Part II of his three-story investigation of the credit crisis, Money Morning Contributing Editor Shah Gilani shows us how American International Group, a perfectly sound company that’s survived for 89 years, was destroyed by some errant bets on a derivative security called a “credit default swap,” or CDS. It’s
a story you’ll read nowhere else.]
By Shah Gilani
Contributing Editor
There’s nothing fundamentally wrong with the core insurance business units of American International Group Inc. (AIG). Nothing at all. What imploded the venerable insurance giant was an accumulation of misplaced bets on credit default swaps.
By the best estimates of the International Swaps and Derivatives Association and the Bank for International Settlements (BIS), often referred to as the central banks’ central bank, the notional value of credit default swaps out in the market place is some $62 trillion, or 35 trillion British Pounds at an exchange rate of $1.78.
A credit default swap (CDS) is akin to an insurance policy. It’s a financial derivative that a debt holder can use to hedge against the default by a debtor corporation of sovereign. But a CDS can also be used to speculate.
A subsidiary of AIG wrote insurance in the form of credit default swaps, meaning it offered buyers insurance protection against losses on debts and loans of borrowers, to the tune of $447 billion. But the mix was toxic. They also sold insurance on esoteric asset-backed security pools – securities like collateralized debt obligations (CDOs), pools of subprime mortgages, pools of Alt-A mortgages, prime mortgage pools and collateralized loan obligations. The subsidiary collected a lot of premium income and its earnings were robust.
When the housing market collapsed, imploding home prices resulted in precipitously rising foreclosures. The mortgage pools AIG insured began to fall in value. Additionally, the credit crisis began to take its toll on leveraged loans and it saw mounting losses on the loan pools it had insured. In 2007, the company was starting to feel serious heat.
From its humble beginnings in China in 1919 – through the 40-year tenure of CEO Maurice R. “Hank” Greenberg, which ended ignominiously for Greenberg in 2006 – AIG grew aggressively. Greenberg grew and diversified the insurance giant, ultimately amassing a trillion-dollar balance sheet.
But not everything was Kosher.
In an effort to assuage analysts and maintain leverage, the firm entered into sham transactions to affect the appearance on its balance sheet of $500 million of loan-loss reserves, which analysts had been questioning as formerly declining. The result was a 2006 Securities and Exchange Commission enforcement action, a $1.6 billion settlement and the removal of Greenberg. Greenberg is still fighting civil charges related to his actions at the firm.
As 2007 progressed, so did the losses on AIG’s books and credit default swaps. Once again, it appears that AIG tried to “manage” the problem through accounting maneuvers. Last February, for instance, AIG said that “its auditor had found a material weakness in its accounting.” It had not been properly valuing its CDO liabilities and swap-related write-downs. The losses were revealed to be in excess of $20 billion through this year’s first quarter. The SEC is once again investigating, as are criminal prosecutors at the U.S. Justice Department and the U.S. Attorney’s Office in Brooklyn.
After writing down assets against gains elsewhere, AIG posted cumulative losses of $18 billion over the last three quarters. In February, AIG posted $5.3 billion in collateral against credit default swap contracts it had written. In April, AIG had to post an additional $4.4 billion in collateral. When rating agencies Standard & Poor’s, Moody’s Investors Service (MCO) and Fitch Ratings Inc., lowered the firm’s ratings last Monday evening, it triggered an additional $14 billion collateral call as margin against AIG’s credit default swaps.
The company didn’t have the cash.
Indeed, the dire need for cash collateral on top of mounting losses on warehoused CDO “assets” on the company’s balance sheet necessitated a massive infusion of capital. That’s what happened to AIG.
But once again, there’s the story – and there’s the story behind the story.
There’s a problem – an inherently systemic problem – and it has to do with how structured investments like tranched collateralized debt obligations (CDOs), residential mortgage-backed securities (RMBS), commercial mortgage-backed securities (CMBS), and credit default swaps on them and on corporate debts and loans are actually valued.
Individually, CDOs are hard to value. Suffice it to say, legend has it that constructing the cash flow payments on the first theoretical 3-tranche CDO (the simplest type of CDO) took a Cray Inc. (CRAY) supercomputer 48 hours. Now try and value credit default swaps on them!
Because there are so many different individual CDO securities, and because there are so many credit default swaps on so many of these CDOs, and so many swaps on individually referenced entity debts and loans, the only way to value them in a portfolio is by indexing.
That’s right, there are indexes, and guess what? You can trade the indexes! Markit Group Ltd., of London, constructs and manages the CDX, ABX, CMBX and LCDX family of credit-default-swap indexes. Investopedia has a decent little tutorial.
Here’s the problem: If you own a portfolio of CDOs, and the only way to value them (or, at least, to develop a valuation that others are reasonably certain to respect), is by looking at them through the prism of an index of credit default swaps on them, you’re at the mercy of the index. Your portfolio, your securities may not be so bad, but you may not really know based on mortgage-duration analysis and foreclosure events that you can’t calculate. So you value, or mark-to-market, against the closest index.
Here’s the rub. What if other speculators are selling short – that is, betting in anticipation of that index going down? What if large portfolio-hedgers are selling short the index to hedge the portfolio they can’t sell because no one will buy it – because no one knows what it’s worth?
It’s crazy. And it gets worse.
What if you’re running a profitable company that needs to borrow money, but credit default swaps (bets against your ability to pay back your debt) are expensive by virtue of speculators fear and greed, such that if any bank looks at where the CDS pricing on your paper is trading, they tell you: “Sorry, but we can’t lend you money because the market for credit default swaps thinks you’re a bad bet.”
You don’t get the loan. You can’t build your factory; you can’t produce and have nothing to sell. The upshot: Now you actually are going out of business. Is this self-fulfilling?
Ponder this: Last Monday, as AIG was initially seeking $20 billion in capital and actually had it in hand (by virtue of a deal with New York insurance regulators), traders were bidding up credit default swaps on AIG’s debt and loans so furiously that based on the insurance premiums traders were actually paying for default insurance on AIG… the company was already dead. Self-fulfilling?
Credit default swaps are creating a downward spiral in the capital markets, driving up the cost of capital, and squeezing out all manner of borrowers. And these speculative bets run amok are undermining all U.S. Federal Reserve and U.S. Treasury Department efforts to “liquefy” the system. If this keeps up, the credit default market could sink the U.S. economy into a recession/depression that will make the Great Depression look like a day at the beach.
Anyone got a towel?
mfg ipollit
Monday, September 22nd, 2008
The Credit Crisis and the Real Story Behind the Collapse of AIG
[In Part II of his three-story investigation of the credit crisis, Money Morning Contributing Editor Shah Gilani shows us how American International Group, a perfectly sound company that’s survived for 89 years, was destroyed by some errant bets on a derivative security called a “credit default swap,” or CDS. It’s
a story you’ll read nowhere else.]
By Shah Gilani
Contributing Editor
There’s nothing fundamentally wrong with the core insurance business units of American International Group Inc. (AIG). Nothing at all. What imploded the venerable insurance giant was an accumulation of misplaced bets on credit default swaps.
By the best estimates of the International Swaps and Derivatives Association and the Bank for International Settlements (BIS), often referred to as the central banks’ central bank, the notional value of credit default swaps out in the market place is some $62 trillion, or 35 trillion British Pounds at an exchange rate of $1.78.
A credit default swap (CDS) is akin to an insurance policy. It’s a financial derivative that a debt holder can use to hedge against the default by a debtor corporation of sovereign. But a CDS can also be used to speculate.
A subsidiary of AIG wrote insurance in the form of credit default swaps, meaning it offered buyers insurance protection against losses on debts and loans of borrowers, to the tune of $447 billion. But the mix was toxic. They also sold insurance on esoteric asset-backed security pools – securities like collateralized debt obligations (CDOs), pools of subprime mortgages, pools of Alt-A mortgages, prime mortgage pools and collateralized loan obligations. The subsidiary collected a lot of premium income and its earnings were robust.
When the housing market collapsed, imploding home prices resulted in precipitously rising foreclosures. The mortgage pools AIG insured began to fall in value. Additionally, the credit crisis began to take its toll on leveraged loans and it saw mounting losses on the loan pools it had insured. In 2007, the company was starting to feel serious heat.
From its humble beginnings in China in 1919 – through the 40-year tenure of CEO Maurice R. “Hank” Greenberg, which ended ignominiously for Greenberg in 2006 – AIG grew aggressively. Greenberg grew and diversified the insurance giant, ultimately amassing a trillion-dollar balance sheet.
But not everything was Kosher.
In an effort to assuage analysts and maintain leverage, the firm entered into sham transactions to affect the appearance on its balance sheet of $500 million of loan-loss reserves, which analysts had been questioning as formerly declining. The result was a 2006 Securities and Exchange Commission enforcement action, a $1.6 billion settlement and the removal of Greenberg. Greenberg is still fighting civil charges related to his actions at the firm.
As 2007 progressed, so did the losses on AIG’s books and credit default swaps. Once again, it appears that AIG tried to “manage” the problem through accounting maneuvers. Last February, for instance, AIG said that “its auditor had found a material weakness in its accounting.” It had not been properly valuing its CDO liabilities and swap-related write-downs. The losses were revealed to be in excess of $20 billion through this year’s first quarter. The SEC is once again investigating, as are criminal prosecutors at the U.S. Justice Department and the U.S. Attorney’s Office in Brooklyn.
After writing down assets against gains elsewhere, AIG posted cumulative losses of $18 billion over the last three quarters. In February, AIG posted $5.3 billion in collateral against credit default swap contracts it had written. In April, AIG had to post an additional $4.4 billion in collateral. When rating agencies Standard & Poor’s, Moody’s Investors Service (MCO) and Fitch Ratings Inc., lowered the firm’s ratings last Monday evening, it triggered an additional $14 billion collateral call as margin against AIG’s credit default swaps.
The company didn’t have the cash.
Indeed, the dire need for cash collateral on top of mounting losses on warehoused CDO “assets” on the company’s balance sheet necessitated a massive infusion of capital. That’s what happened to AIG.
But once again, there’s the story – and there’s the story behind the story.
There’s a problem – an inherently systemic problem – and it has to do with how structured investments like tranched collateralized debt obligations (CDOs), residential mortgage-backed securities (RMBS), commercial mortgage-backed securities (CMBS), and credit default swaps on them and on corporate debts and loans are actually valued.
Individually, CDOs are hard to value. Suffice it to say, legend has it that constructing the cash flow payments on the first theoretical 3-tranche CDO (the simplest type of CDO) took a Cray Inc. (CRAY) supercomputer 48 hours. Now try and value credit default swaps on them!
Because there are so many different individual CDO securities, and because there are so many credit default swaps on so many of these CDOs, and so many swaps on individually referenced entity debts and loans, the only way to value them in a portfolio is by indexing.
That’s right, there are indexes, and guess what? You can trade the indexes! Markit Group Ltd., of London, constructs and manages the CDX, ABX, CMBX and LCDX family of credit-default-swap indexes. Investopedia has a decent little tutorial.
Here’s the problem: If you own a portfolio of CDOs, and the only way to value them (or, at least, to develop a valuation that others are reasonably certain to respect), is by looking at them through the prism of an index of credit default swaps on them, you’re at the mercy of the index. Your portfolio, your securities may not be so bad, but you may not really know based on mortgage-duration analysis and foreclosure events that you can’t calculate. So you value, or mark-to-market, against the closest index.
Here’s the rub. What if other speculators are selling short – that is, betting in anticipation of that index going down? What if large portfolio-hedgers are selling short the index to hedge the portfolio they can’t sell because no one will buy it – because no one knows what it’s worth?
It’s crazy. And it gets worse.
What if you’re running a profitable company that needs to borrow money, but credit default swaps (bets against your ability to pay back your debt) are expensive by virtue of speculators fear and greed, such that if any bank looks at where the CDS pricing on your paper is trading, they tell you: “Sorry, but we can’t lend you money because the market for credit default swaps thinks you’re a bad bet.”
You don’t get the loan. You can’t build your factory; you can’t produce and have nothing to sell. The upshot: Now you actually are going out of business. Is this self-fulfilling?
Ponder this: Last Monday, as AIG was initially seeking $20 billion in capital and actually had it in hand (by virtue of a deal with New York insurance regulators), traders were bidding up credit default swaps on AIG’s debt and loans so furiously that based on the insurance premiums traders were actually paying for default insurance on AIG… the company was already dead. Self-fulfilling?
Credit default swaps are creating a downward spiral in the capital markets, driving up the cost of capital, and squeezing out all manner of borrowers. And these speculative bets run amok are undermining all U.S. Federal Reserve and U.S. Treasury Department efforts to “liquefy” the system. If this keeps up, the credit default market could sink the U.S. economy into a recession/depression that will make the Great Depression look like a day at the beach.
Anyone got a towel?
mfg ipollit
Antwort auf Beitrag Nr.: 35.395.860 von ipollit am 03.10.08 13:36:15http://www.moneymorning.com/2008/09/24/financial-meltdown/
Wednesday, September 24th, 2008
How Complex Securities, Wall Street Protectionism and Myopic Regulation Caused a Near-Meltdown of the U.S. Banking System
[In Part III of his three-story investigation of the credit crisis,Money MorningContributing Editor Shah Gilani details how the very complexity of the global financial system brought us to the brink of a total meltdown. In a special addendum tomorrow (Thursday), the former professional trader and hedge-fund manager will detail a banking-system overhaul that would immediately end the credit crisis - possibly without a single penny of taxpayer money.]
By Shah Gilani
Contributing Editor
There’s no time to beat around the bush. Let’s flush out the three credit-crisis catalysts that have remained hidden for too long, thanks to Wall Street protectionism and myopic regulation. Those catalysts - which brought us to the brink of a financial meltdown - are structured collateralized debt obligations, credit default swaps, and the horrific offspring of the two - credit default swaps on structured collateralized debt obligations.
An asset-backed security (ABS) is a type of tradable debt security that’s derived from a pool of underlying assets. We could be talking about a pool of mortgages, of automobile leases, or loans made to various borrowers. We’re using the example of residential mortgages, though the example is exactly the same for commercial mortgages, automobile leases or bank loans. Here’s how it works.
Anatomy of Mortgage Loan
A mortgage company makes home loans in your county, as does your local bank branch. Then an investment bank comes along and buys the mortgages from the mortgage company and from the bank. It only wants to buy the mortgages made to prime borrowers who are paying 6% interest on their mortgages. Once it acquires those loans, the investment bank securitizes the mortgages, meaning it pools them into a tradable package it can sell to investors.
This particular pool is known as a "closed pool," meaning no more mortgages will be added, though some may leave the pool if the underlying borrowers pay back their mortgages early because they sold their homes, or refinanced them, or if underlying mortgages are in default and the "servicer" allows them to be removed from the pool. The only income coming into the closed pool results from the monthly interest and principal payments being made by the homeowners.
In our example - because all the mortgage loans were made to so-called "prime" borrowers with strong credit - you might have an investment grade (A+) security that pays 6%, because all the mortgage holders are paying 6% and the payments are being passed through to the investors. That’s it. There are very good, though not exact, methodologies to value this particular security, primarily because it is uniform in that all the mortgage payers are prime borrowers who all are paying 6%.
Asset-backed-securities become infinitely more complicated when they are sliced and diced into structured collateralized instruments. They generally fit into two main categories:
Collateralized debt obligations (CDOs), which include all manner of residential and commercial mortgage-backed securities.
And collateralized loan obligations (CLOs), which are pooled bank and investment-bank loan portfolios.
CDOs and CLOs are created from "closed-pool," asset-backed securities. They are collateralized by the underlying assets - hence the prefix - but they are also "structured."
In our example above, our asset-backed mortgage security was rated A+ and pays the investor who buys it 6%. If I want to create higher-yielding securities that I think I will be able to sell a lot more of, I will pool mortgages from subprime borrowers.
Because subprime borrowers are, by definition, higher-risk borrowers, the mortgage companies and banks charge them higher rates of interest to offset the greater risk that they represent. If I pool these mortgages, their ratings would be "junk" - or close to it - which will be a problem as I try and sell these securities to investors all around the world.
That’s where the magic of financial engineering, better known as structuring, comes into play. I can divide up the closed pool of subprime mortgages and structure the pool into layers, or tranches. What I’ll do is divide up the pool into multiple tranches, or slices. I’ll structure the cash flow payments from all the mortgages so that if the 1st or 2nd tranches run into trouble, I’ll take cash flow payments from the lower tranches to keep up with all the payments to the holders of the 1st and 2nd tranches.
For someone trying to peddle these asset-backed securities, this is a stroke of genius. In our example, since I’m now pretty much guaranteeing that the 1st and 2nd tranche security holders are going to get paid, maybe I can get the Big Three debt-rating companies - Standard & Poor’s, Moody’s Investors Service (MCO) and Fitch Ratings Inc. - to give my 1st and 2nd tranche CDOs’ investment grade ratings. Maybe I can even buy insurance from a monoline insurer like AMBAC Financial Group Inc. (ABK) or MBIA Inc. (MBIA), and get my top tranches a coveted "AAA" rating. Wow, I could sure sell a lot of this high-yielding stuff with an investment grade rating!
That’s just what happened. And they did sell a lot - a whole lot.
Those Troubling Tranches
As I said in Part II of this investigative series, CDOs - on an individual basis - are difficult to value. Indeed, "legend has it that constructing the cash flow payments on the first theoretical 3-tranche CDO (the simplest type of CDO) took a Cray Inc. (CRAY) supercomputer 48 hours to calculate.
The problem starts here. There are so many of these tranched securities out in the marketplace - and on the balance sheets of banks, investment banks, insurance companies, hedge funds and all manner of other unsuspecting investment entities worldwide - that when subprime borrowers began to default, it wasn’t long before the lower-tier tranches ran out of money to pay the so-called 1st- and 2nd-tier "AAA"-rated securities. The problem escalated quickly and almost all of these securities were downgraded. That’s not a surprise. Nor is it the whole story, for it leaves a key question unanswered.
What happened to the lowest-level tranches?
Those tranches were "ugly" to begin with because I started by pooling subprime mortgages (the high-risk borrowers). Then I made them "toxic" by "stripping out" their cash flow to support other tranches. This toxic waste was so bad, no one would ever rate it and only greedy hedge funds or crazy speculators would buy it for its high yield. Or, maybe, I think so much of my creation that I’ll keep this piece for myself, or maybe I’ll have to because no investor will ever buy it.
This kind of stuff is out there. There’s a lot of it. And only an act of God will bring these securities back from the depths where they now reside.
With their collateralized premise and structured nature, CDOs are very difficult to value - especially since no one trusts anyone else’s "internal valuation model." Since everyone is afraid of these securities because no one really knows what they’re actually worth, no one wants to buy them.
However, when an institution - such as a Merrill Lynch & Co. Inc. (MER) - gets desperate enough to sell a portfolio of these securities at 22 cents on the dollar, then everyone else who has to "mark-to-market" their assets now has to value similar securities of their own at 22 cents on the dollar. That causes massive write-downs at banks, investment banks, insurance companies, and other financial institutions. And these companies write down assets and watch their losses escalate, they are forced to raise additional capital to meet regulatory requirements.
CDS - Controlled Dangerous (Financial) Substances
It’s a vicious cycle - one that’s eroding our faith in our banks, and worse, banks’ faith in other banks. As a result, banks have ceased lending to each other out of the fear that the next round of write-downs and losses may imperil some of the trading partner banks that they used to lend billions of dollars to every night.
Not anymore.
It would be bad enough if that were the only problem facing the securities market. On top of these overly engineered structured securities I’ve just discussed, we also have credit default swaps with an estimated notional value of $62 trillion out in the marketplace. A credit default swap (CDS) is a financial derivative that’s akin to an insurance policy that a debt holder can use to hedge against the default by a debtor corporation, or a sovereign entity. But a CDS can also be used to speculate.
In Part II of our investigation, which ran Monday, I explained how problematic credit default swap pricing is and how the indexes against which the value of these swaps are determined are tradable themselves as speculative instruments and how the whole complex is driving the financial system into an abyss. That’s essentially what led to the collapse of the otherwise healthy insurance giant, American International Group Inc. (AIG). [For the latest news on AIG, check out this related story elsewhere in today's issue of Money Morning.]
Unfortunately, I don’t see the U.S. Treasury Department’s much-needed rescue plan being effective without actually addressing the problems facing both the CDO and the CDS markets. The Treasury Department’s initiative will create more problems than they attempt to solve and will eventually saddle taxpayers with so much debt that they risk sinking the dollar, and worse, the U.S. government’s investment grade rating. That would be calamitous. [For the latest news on the federal government's banking-system bailout plan, check out this related story elsewhere in today's issue of Money Morning.]
Tomorrow (Thursday) in Money Morning, in an addendum to this piece, I will outline a proposal that I’m calling the Money Morning Plan because it potentially heralds a new dawn in the credit crisis, addressing the problems from the bottom up, and not from the top down. Although this plan is straightforward and elegant in its simplicity, we still opted to present it as a separate story in order to provide you with the focus, the detail and the explanations we feel this strategy merits.
If the Treasury Department wants to immediately triage the gushing wounds that are bleeding our banks and financial system dry of readily available credit by purchasing and warehousing illiquid assets with taxpayer money, it won’t be long before the U.S. financial system begins to hemorrhage somewhere else.
The free market caused these problems under the noses of undistinguished regulators.
The free market - with the oversight of good governance practices mandated by effective regulators, who should not be empowered to kill entrepreneurial capitalism - will once again rise to the occasion and prove America’s robustness and indefatigable spirit.
mfg ipollit
Wednesday, September 24th, 2008
How Complex Securities, Wall Street Protectionism and Myopic Regulation Caused a Near-Meltdown of the U.S. Banking System
[In Part III of his three-story investigation of the credit crisis,Money MorningContributing Editor Shah Gilani details how the very complexity of the global financial system brought us to the brink of a total meltdown. In a special addendum tomorrow (Thursday), the former professional trader and hedge-fund manager will detail a banking-system overhaul that would immediately end the credit crisis - possibly without a single penny of taxpayer money.]
By Shah Gilani
Contributing Editor
There’s no time to beat around the bush. Let’s flush out the three credit-crisis catalysts that have remained hidden for too long, thanks to Wall Street protectionism and myopic regulation. Those catalysts - which brought us to the brink of a financial meltdown - are structured collateralized debt obligations, credit default swaps, and the horrific offspring of the two - credit default swaps on structured collateralized debt obligations.
An asset-backed security (ABS) is a type of tradable debt security that’s derived from a pool of underlying assets. We could be talking about a pool of mortgages, of automobile leases, or loans made to various borrowers. We’re using the example of residential mortgages, though the example is exactly the same for commercial mortgages, automobile leases or bank loans. Here’s how it works.
Anatomy of Mortgage Loan
A mortgage company makes home loans in your county, as does your local bank branch. Then an investment bank comes along and buys the mortgages from the mortgage company and from the bank. It only wants to buy the mortgages made to prime borrowers who are paying 6% interest on their mortgages. Once it acquires those loans, the investment bank securitizes the mortgages, meaning it pools them into a tradable package it can sell to investors.
This particular pool is known as a "closed pool," meaning no more mortgages will be added, though some may leave the pool if the underlying borrowers pay back their mortgages early because they sold their homes, or refinanced them, or if underlying mortgages are in default and the "servicer" allows them to be removed from the pool. The only income coming into the closed pool results from the monthly interest and principal payments being made by the homeowners.
In our example - because all the mortgage loans were made to so-called "prime" borrowers with strong credit - you might have an investment grade (A+) security that pays 6%, because all the mortgage holders are paying 6% and the payments are being passed through to the investors. That’s it. There are very good, though not exact, methodologies to value this particular security, primarily because it is uniform in that all the mortgage payers are prime borrowers who all are paying 6%.
Asset-backed-securities become infinitely more complicated when they are sliced and diced into structured collateralized instruments. They generally fit into two main categories:
Collateralized debt obligations (CDOs), which include all manner of residential and commercial mortgage-backed securities.
And collateralized loan obligations (CLOs), which are pooled bank and investment-bank loan portfolios.
CDOs and CLOs are created from "closed-pool," asset-backed securities. They are collateralized by the underlying assets - hence the prefix - but they are also "structured."
In our example above, our asset-backed mortgage security was rated A+ and pays the investor who buys it 6%. If I want to create higher-yielding securities that I think I will be able to sell a lot more of, I will pool mortgages from subprime borrowers.
Because subprime borrowers are, by definition, higher-risk borrowers, the mortgage companies and banks charge them higher rates of interest to offset the greater risk that they represent. If I pool these mortgages, their ratings would be "junk" - or close to it - which will be a problem as I try and sell these securities to investors all around the world.
That’s where the magic of financial engineering, better known as structuring, comes into play. I can divide up the closed pool of subprime mortgages and structure the pool into layers, or tranches. What I’ll do is divide up the pool into multiple tranches, or slices. I’ll structure the cash flow payments from all the mortgages so that if the 1st or 2nd tranches run into trouble, I’ll take cash flow payments from the lower tranches to keep up with all the payments to the holders of the 1st and 2nd tranches.
For someone trying to peddle these asset-backed securities, this is a stroke of genius. In our example, since I’m now pretty much guaranteeing that the 1st and 2nd tranche security holders are going to get paid, maybe I can get the Big Three debt-rating companies - Standard & Poor’s, Moody’s Investors Service (MCO) and Fitch Ratings Inc. - to give my 1st and 2nd tranche CDOs’ investment grade ratings. Maybe I can even buy insurance from a monoline insurer like AMBAC Financial Group Inc. (ABK) or MBIA Inc. (MBIA), and get my top tranches a coveted "AAA" rating. Wow, I could sure sell a lot of this high-yielding stuff with an investment grade rating!
That’s just what happened. And they did sell a lot - a whole lot.
Those Troubling Tranches
As I said in Part II of this investigative series, CDOs - on an individual basis - are difficult to value. Indeed, "legend has it that constructing the cash flow payments on the first theoretical 3-tranche CDO (the simplest type of CDO) took a Cray Inc. (CRAY) supercomputer 48 hours to calculate.
The problem starts here. There are so many of these tranched securities out in the marketplace - and on the balance sheets of banks, investment banks, insurance companies, hedge funds and all manner of other unsuspecting investment entities worldwide - that when subprime borrowers began to default, it wasn’t long before the lower-tier tranches ran out of money to pay the so-called 1st- and 2nd-tier "AAA"-rated securities. The problem escalated quickly and almost all of these securities were downgraded. That’s not a surprise. Nor is it the whole story, for it leaves a key question unanswered.
What happened to the lowest-level tranches?
Those tranches were "ugly" to begin with because I started by pooling subprime mortgages (the high-risk borrowers). Then I made them "toxic" by "stripping out" their cash flow to support other tranches. This toxic waste was so bad, no one would ever rate it and only greedy hedge funds or crazy speculators would buy it for its high yield. Or, maybe, I think so much of my creation that I’ll keep this piece for myself, or maybe I’ll have to because no investor will ever buy it.
This kind of stuff is out there. There’s a lot of it. And only an act of God will bring these securities back from the depths where they now reside.
With their collateralized premise and structured nature, CDOs are very difficult to value - especially since no one trusts anyone else’s "internal valuation model." Since everyone is afraid of these securities because no one really knows what they’re actually worth, no one wants to buy them.
However, when an institution - such as a Merrill Lynch & Co. Inc. (MER) - gets desperate enough to sell a portfolio of these securities at 22 cents on the dollar, then everyone else who has to "mark-to-market" their assets now has to value similar securities of their own at 22 cents on the dollar. That causes massive write-downs at banks, investment banks, insurance companies, and other financial institutions. And these companies write down assets and watch their losses escalate, they are forced to raise additional capital to meet regulatory requirements.
CDS - Controlled Dangerous (Financial) Substances
It’s a vicious cycle - one that’s eroding our faith in our banks, and worse, banks’ faith in other banks. As a result, banks have ceased lending to each other out of the fear that the next round of write-downs and losses may imperil some of the trading partner banks that they used to lend billions of dollars to every night.
Not anymore.
It would be bad enough if that were the only problem facing the securities market. On top of these overly engineered structured securities I’ve just discussed, we also have credit default swaps with an estimated notional value of $62 trillion out in the marketplace. A credit default swap (CDS) is a financial derivative that’s akin to an insurance policy that a debt holder can use to hedge against the default by a debtor corporation, or a sovereign entity. But a CDS can also be used to speculate.
In Part II of our investigation, which ran Monday, I explained how problematic credit default swap pricing is and how the indexes against which the value of these swaps are determined are tradable themselves as speculative instruments and how the whole complex is driving the financial system into an abyss. That’s essentially what led to the collapse of the otherwise healthy insurance giant, American International Group Inc. (AIG). [For the latest news on AIG, check out this related story elsewhere in today's issue of Money Morning.]
Unfortunately, I don’t see the U.S. Treasury Department’s much-needed rescue plan being effective without actually addressing the problems facing both the CDO and the CDS markets. The Treasury Department’s initiative will create more problems than they attempt to solve and will eventually saddle taxpayers with so much debt that they risk sinking the dollar, and worse, the U.S. government’s investment grade rating. That would be calamitous. [For the latest news on the federal government's banking-system bailout plan, check out this related story elsewhere in today's issue of Money Morning.]
Tomorrow (Thursday) in Money Morning, in an addendum to this piece, I will outline a proposal that I’m calling the Money Morning Plan because it potentially heralds a new dawn in the credit crisis, addressing the problems from the bottom up, and not from the top down. Although this plan is straightforward and elegant in its simplicity, we still opted to present it as a separate story in order to provide you with the focus, the detail and the explanations we feel this strategy merits.
If the Treasury Department wants to immediately triage the gushing wounds that are bleeding our banks and financial system dry of readily available credit by purchasing and warehousing illiquid assets with taxpayer money, it won’t be long before the U.S. financial system begins to hemorrhage somewhere else.
The free market caused these problems under the noses of undistinguished regulators.
The free market - with the oversight of good governance practices mandated by effective regulators, who should not be empowered to kill entrepreneurial capitalism - will once again rise to the occasion and prove America’s robustness and indefatigable spirit.
mfg ipollit
Eli Lilly hat BMS ausgestochen und sich mit Imclone auf eine Übernahme für 70 USD pro Aktie geeinigt. Es ist etwas seltsam, dass jemand anderes außer BMS oder Merck an IMCL Interesse hat, da die beiden ersten einen großen Teil der Rechte an Imclone's einzigem zugelassenen Mittel Erbitux und größere Teile vom Unternehmen selbst halten. Der Preis muss sich demnach aus der Pipeline ergeben. BMS kann nur noch etwas machen mit dem Versuch einer feindlichen Übernahme zu einem höheren Preis als 70 USD pps...
vielleicht kann Genmab's Zalutumumab Erbitux bald arg in Bedrängnis bringen, wenn im März die PIII-Daten sehr gut aussehen.
06.10.2008 13:12
ImClone Announces Merger Agreement with Eli Lilly at $70 Per Share
ImClone Systems Incorporated (News)
(NASDAQ: IMCL) announced today that it has entered into a merger agreement with Eli Lilly and Company (News/Aktienkurs) (NYSE: LLY), pursuant to which Lilly has agreed to commence a tender offer for no less than a majority of the issued and outstanding shares of ImClone common stock at a net price per share of $70 in cash. Assuming that the tender offer is successful, the merger agreement provides that the tender offer will be followed by a merger pursuant to which ImClone would be acquired by Lilly. ImClone stockholders who did not tender would receive the same consideration as the tendering stockholders. This transaction represents a premium of 51% to the closing price of $46.44 per share on July 30, 2008, one day prior to the announcement of Bristol-Myers Squibb's (”BMS“) offer for ImClone and represents a $10 premium to BMS's offer of $60 and an $8 premium to BMS's proposed tender offer price of $62.
In addition to the minimum tender requirement, the transaction is subject to customary closing conditions, including regulatory approvals.
Carl Icahn, Chairman of ImClone's Board of Directors stated: ”We are extremely pleased to be able to present our stockholders with an offer that, if accepted, will bring them $70 per share in cash. I would like to thank the stockholders who have been supportive of me and the new directors who replaced the old regime.“
”We came on board after a proxy contest and consent solicitation in which we criticized the old regime. Since then we were able to cut costs, substantially improve our relationship with Bristol-Myers Squibb and continue the expansion of the use of ERBITUX® in fighting certain cancers while developing what is a promising pipeline.“
”We feel that the Eli Lilly transaction vindicates our decision to oppose in 2006 a potential transaction in which the Company would have been sold at approximately $36 per share which the prior board favored.“
”Finally, I want to thank my fellow Board members and ImClone employees for their help in bringing this transaction to fruition. I especially want to acknowledge the roles of Alex Denner and Richard Mulligan, two Board members, for their yeoman service to ImClone after the proxy contest to help turn the Company around.“ Mr. Icahn concluded by stating that ”while it is easy to hurl stones, all stockholders owe a debt of gratitude to Sam Waksal without whose dedication and perseverance neither ERBITUX nor our great pipeline would exist.“
John H. Johnson, ImClone's Chief Executive Officer, said, ”This combination delivers compelling and certain value to ImClone stockholders.“
mfg ipollit
vielleicht kann Genmab's Zalutumumab Erbitux bald arg in Bedrängnis bringen, wenn im März die PIII-Daten sehr gut aussehen.
06.10.2008 13:12
ImClone Announces Merger Agreement with Eli Lilly at $70 Per Share
ImClone Systems Incorporated (News)
(NASDAQ: IMCL) announced today that it has entered into a merger agreement with Eli Lilly and Company (News/Aktienkurs) (NYSE: LLY), pursuant to which Lilly has agreed to commence a tender offer for no less than a majority of the issued and outstanding shares of ImClone common stock at a net price per share of $70 in cash. Assuming that the tender offer is successful, the merger agreement provides that the tender offer will be followed by a merger pursuant to which ImClone would be acquired by Lilly. ImClone stockholders who did not tender would receive the same consideration as the tendering stockholders. This transaction represents a premium of 51% to the closing price of $46.44 per share on July 30, 2008, one day prior to the announcement of Bristol-Myers Squibb's (”BMS“) offer for ImClone and represents a $10 premium to BMS's offer of $60 and an $8 premium to BMS's proposed tender offer price of $62.
In addition to the minimum tender requirement, the transaction is subject to customary closing conditions, including regulatory approvals.
Carl Icahn, Chairman of ImClone's Board of Directors stated: ”We are extremely pleased to be able to present our stockholders with an offer that, if accepted, will bring them $70 per share in cash. I would like to thank the stockholders who have been supportive of me and the new directors who replaced the old regime.“
”We came on board after a proxy contest and consent solicitation in which we criticized the old regime. Since then we were able to cut costs, substantially improve our relationship with Bristol-Myers Squibb and continue the expansion of the use of ERBITUX® in fighting certain cancers while developing what is a promising pipeline.“
”We feel that the Eli Lilly transaction vindicates our decision to oppose in 2006 a potential transaction in which the Company would have been sold at approximately $36 per share which the prior board favored.“
”Finally, I want to thank my fellow Board members and ImClone employees for their help in bringing this transaction to fruition. I especially want to acknowledge the roles of Alex Denner and Richard Mulligan, two Board members, for their yeoman service to ImClone after the proxy contest to help turn the Company around.“ Mr. Icahn concluded by stating that ”while it is easy to hurl stones, all stockholders owe a debt of gratitude to Sam Waksal without whose dedication and perseverance neither ERBITUX nor our great pipeline would exist.“
John H. Johnson, ImClone's Chief Executive Officer, said, ”This combination delivers compelling and certain value to ImClone stockholders.“
mfg ipollit
OSIP... Traceva+Avastin Kombination in NSLC wieder gescheitert... beide für sich alleine sind bereits zugelassen. Avastin bringt gegenüber Traceva alleine keine Verbesserung.
Genentech and OSI Pharmaceuticals Announce Topline Results from Phase III Study Evaluating the Combination of Avastin and Tarceva as Second-Line Treatment for Advanced Non-Small Cell Lung Cancer
Monday October 6, 1:30 am ET
SOUTH SAN FRANCISCO, Calif. & MELVILLE, N.Y.--(BUSINESS WIRE)--Genentech, Inc. (NYSE: DNA - News) and OSI Pharmaceuticals, Inc. (Nasdaq: OSIP - News) today announced that a randomized Phase III study (BeTa Lung) evaluating Avastin® (bevacizumab) in combination with Tarceva® (erlotinib) in patients with advanced non-small cell lung cancer (NSCLC) whose disease had progressed following platinum-based chemotherapy did not meet its primary endpoint of improving overall survival compared to Tarceva in combination with placebo. However, there was clear evidence of clinical activity with improvements in the secondary endpoints of progression-free survival (PFS) and response rate when Avastin was added to Tarceva compared to Tarceva alone in this study.
Median survival was similar in both arms of BeTa Lung. No new or unexpected safety signals for either Avastin or Tarceva were observed in the study, and adverse events were consistent with those observed in previous NSCLC clinical trials evaluating the agents. The companies are further analyzing the study results and will submit the data for presentation at the 2008 Chicago Multidisciplinary Symposium in Thoracic Oncology in Chicago, Ill., November 13-15.
mfg ipollit
Genentech and OSI Pharmaceuticals Announce Topline Results from Phase III Study Evaluating the Combination of Avastin and Tarceva as Second-Line Treatment for Advanced Non-Small Cell Lung Cancer
Monday October 6, 1:30 am ET
SOUTH SAN FRANCISCO, Calif. & MELVILLE, N.Y.--(BUSINESS WIRE)--Genentech, Inc. (NYSE: DNA - News) and OSI Pharmaceuticals, Inc. (Nasdaq: OSIP - News) today announced that a randomized Phase III study (BeTa Lung) evaluating Avastin® (bevacizumab) in combination with Tarceva® (erlotinib) in patients with advanced non-small cell lung cancer (NSCLC) whose disease had progressed following platinum-based chemotherapy did not meet its primary endpoint of improving overall survival compared to Tarceva in combination with placebo. However, there was clear evidence of clinical activity with improvements in the secondary endpoints of progression-free survival (PFS) and response rate when Avastin was added to Tarceva compared to Tarceva alone in this study.
Median survival was similar in both arms of BeTa Lung. No new or unexpected safety signals for either Avastin or Tarceva were observed in the study, and adverse events were consistent with those observed in previous NSCLC clinical trials evaluating the agents. The companies are further analyzing the study results and will submit the data for presentation at the 2008 Chicago Multidisciplinary Symposium in Thoracic Oncology in Chicago, Ill., November 13-15.
mfg ipollit
Genmab... PIII von HuMax-CD4 wegen schwachem Kosten/Gewinn-Potential gestoppt. 100 Mitarbeiter entlassen von (ca. 400). HuMax-EGFr Studien gegen Lungen- und Darmkrebs vorerst angehalten, um Ressourcen zu sparen. Konzentration auf Krebs. Angeblich soll dies nichts mit der Finanzkrise zutun haben.
Genmab A/S - Unternehmensmitteilung: Genmab meldet Portfolioergebnisse
KOPENHAGEN, October 9 /PRNewswire/ --
- Zanolimumab-Programm wird nach Prüfung eingestellt, Anzahl der Mitarbeiter wird reduziert
- Zusammenfassung: Genmab meldet die Ergebnisse seiner Portfolioprüfung; Zanolimumab-Programm wird eingestellt und das Unternehmen reduziert die Anzahl seiner Mitarbeiter
Genmab A/S (OMX: GEN) meldet heute die Einstellung der Entwicklung von Zanolimumab (HuMax-CD4(R)). Aufgrund einer Portfolioprüfung und einer Beurteilung der Organisation wird Genmab ausserdem 101 Arbeitsplätze abbauen.
Während sich das Unternehmen einer potenziellen Kommerzialisierung nähert, besteht eine der wesentlichen Prioritäten darin, ein nachhaltiges F&E-Investitionsniveau zu erreichen. Genmab hat eine Prüfung seines Portfolios und seiner Organisation durchgeführt, um Prioritäten zu bestimmen, die den grössten potenziellen Wert bieten. Das Unternehmen wird seinen Fokus auf Arzneimittel zur Krebsbekämpfung verschärfen und sich auf ein weniger breites Portfolio mit einem höheren Potenzial konzentrieren.
Für Zanolimumab werden derzeit Pivot-Studien der Phase III zur Behandlung kutaner T-Zellen-Lymphome (CTCL) durchgeführt. Wie Genmab bereits zuvor angedeutet hatte, läuft die Patientenrekrutierung für die Zanolimumab-Pivotstudie recht langsam. Das Unternehmen ist davon überzeugt, dass dies auf das relativ geringe Marktpotenzial bei CTCL, die Einführung neuer Arzneimittel zur Bekämpfung von CTCL auf dem Markt und zahlreiche konkurrierende klinische Studien zurückzuführen ist. Angesichts dieser Aspekte ist Genmab der Ansicht, dass die erhebliche Investition, die erforderlich wäre, um das Produkt bis hin zur Genehmigung zu entwickeln, keine optimale Verwendung seiner Ressourcen mehr darstellt.
Genmab wird ausserdem versuchen, Lizenzen für drei seiner Entwicklungsprogramme in frühen Phasen, die sich ausserhalb des Fokusbereichs befinden (HuMax-HepC(TM), HuMax-IL8(TM) und HuMax-TAC(TM)), extern zu vergeben.
Darüber hinaus wird Genmab die Frühphasenstudien für Zalutumumab (HuMax-EGFr(TM)) für Kolorektal- und Lungenkrebs einstellen. Diese Entscheidung beruht auf neuen Informationen über die Rolle von K-RAS-Mutationen und entsprechenden Therapien. Genmab wird seine Entwicklung von zwei Studien der Phase III und zwei Studien in früheren Phasen mit Zalutumumab für die Behandlung von Kopf- und Halskrebs fortsetzen.
Genmab wird die Mitarbeiterzahl in all seinen internationalen Standorten reduzieren, um die Anzahl der Programme mit den hierfür erforderlichen Ressourcen abzugleichen. Hiervon werden 101 Mitarbeiter des Unternehmens betroffen sein.
Es wird nicht erwartet, dass sich diese Entscheidungen substanziell auf die Prognose für das laufende Jahr auswirken werden.
"Das Board und die Unternehmensleitung von Genmab bedauern zutiefst die Notwendigkeit, unsere Belegschaft reduzieren zu müssen. Bei allen Personen handelt es sich um geschätzte Mitglieder unseres Teams. Die Änderungen sind jedoch erforderlich, um sicherzustellen, dass wir das korrekte Personalniveau für die geplanten Entwicklungsprogramme haben. Angesichts des aktuellen wirtschaftlichen Umfelds ist die Notwendigkeit, diese Änderungen vorzunehmen, besonders dringlich geworden", sagte Lisa N. Drakeman, Ph.D., Chief Executive Officer von Genmab. "Wir sind all unseren Mitgliedern für ihre Beiträge sehr dankbar und werden unser Bestes tun, denjenigen, die gehen müssen, dabei zu helfen, neue Positionen zu finden. Mit einem breiten Entwicklungsprogramm, das sechs laufende Studien der Phase III umfasst, müssen wir unsere Ausgaben priorisieren, um sicherzustellen, dass wir auch weiterhin effektiv in die Zukunft investieren können."
mfg ipollit
Genmab A/S - Unternehmensmitteilung: Genmab meldet Portfolioergebnisse
KOPENHAGEN, October 9 /PRNewswire/ --
- Zanolimumab-Programm wird nach Prüfung eingestellt, Anzahl der Mitarbeiter wird reduziert
- Zusammenfassung: Genmab meldet die Ergebnisse seiner Portfolioprüfung; Zanolimumab-Programm wird eingestellt und das Unternehmen reduziert die Anzahl seiner Mitarbeiter
Genmab A/S (OMX: GEN) meldet heute die Einstellung der Entwicklung von Zanolimumab (HuMax-CD4(R)). Aufgrund einer Portfolioprüfung und einer Beurteilung der Organisation wird Genmab ausserdem 101 Arbeitsplätze abbauen.
Während sich das Unternehmen einer potenziellen Kommerzialisierung nähert, besteht eine der wesentlichen Prioritäten darin, ein nachhaltiges F&E-Investitionsniveau zu erreichen. Genmab hat eine Prüfung seines Portfolios und seiner Organisation durchgeführt, um Prioritäten zu bestimmen, die den grössten potenziellen Wert bieten. Das Unternehmen wird seinen Fokus auf Arzneimittel zur Krebsbekämpfung verschärfen und sich auf ein weniger breites Portfolio mit einem höheren Potenzial konzentrieren.
Für Zanolimumab werden derzeit Pivot-Studien der Phase III zur Behandlung kutaner T-Zellen-Lymphome (CTCL) durchgeführt. Wie Genmab bereits zuvor angedeutet hatte, läuft die Patientenrekrutierung für die Zanolimumab-Pivotstudie recht langsam. Das Unternehmen ist davon überzeugt, dass dies auf das relativ geringe Marktpotenzial bei CTCL, die Einführung neuer Arzneimittel zur Bekämpfung von CTCL auf dem Markt und zahlreiche konkurrierende klinische Studien zurückzuführen ist. Angesichts dieser Aspekte ist Genmab der Ansicht, dass die erhebliche Investition, die erforderlich wäre, um das Produkt bis hin zur Genehmigung zu entwickeln, keine optimale Verwendung seiner Ressourcen mehr darstellt.
Genmab wird ausserdem versuchen, Lizenzen für drei seiner Entwicklungsprogramme in frühen Phasen, die sich ausserhalb des Fokusbereichs befinden (HuMax-HepC(TM), HuMax-IL8(TM) und HuMax-TAC(TM)), extern zu vergeben.
Darüber hinaus wird Genmab die Frühphasenstudien für Zalutumumab (HuMax-EGFr(TM)) für Kolorektal- und Lungenkrebs einstellen. Diese Entscheidung beruht auf neuen Informationen über die Rolle von K-RAS-Mutationen und entsprechenden Therapien. Genmab wird seine Entwicklung von zwei Studien der Phase III und zwei Studien in früheren Phasen mit Zalutumumab für die Behandlung von Kopf- und Halskrebs fortsetzen.
Genmab wird die Mitarbeiterzahl in all seinen internationalen Standorten reduzieren, um die Anzahl der Programme mit den hierfür erforderlichen Ressourcen abzugleichen. Hiervon werden 101 Mitarbeiter des Unternehmens betroffen sein.
Es wird nicht erwartet, dass sich diese Entscheidungen substanziell auf die Prognose für das laufende Jahr auswirken werden.
"Das Board und die Unternehmensleitung von Genmab bedauern zutiefst die Notwendigkeit, unsere Belegschaft reduzieren zu müssen. Bei allen Personen handelt es sich um geschätzte Mitglieder unseres Teams. Die Änderungen sind jedoch erforderlich, um sicherzustellen, dass wir das korrekte Personalniveau für die geplanten Entwicklungsprogramme haben. Angesichts des aktuellen wirtschaftlichen Umfelds ist die Notwendigkeit, diese Änderungen vorzunehmen, besonders dringlich geworden", sagte Lisa N. Drakeman, Ph.D., Chief Executive Officer von Genmab. "Wir sind all unseren Mitgliedern für ihre Beiträge sehr dankbar und werden unser Bestes tun, denjenigen, die gehen müssen, dabei zu helfen, neue Positionen zu finden. Mit einem breiten Entwicklungsprogramm, das sechs laufende Studien der Phase III umfasst, müssen wir unsere Ausgaben priorisieren, um sicherzustellen, dass wir auch weiterhin effektiv in die Zukunft investieren können."
mfg ipollit
11.10.2008 | 00:01 Uhr
Genmab erreicht Meilenstein bei der Ofatumumab-Kooperation
KOPENHAGEN, Dänemark, October 10 (ots/PRNewswire) - -
Zusammenfassung: Genmab erreicht sechsten Meilenstein in der Ofatumumab-Kooperation mit GSK
Genmab A/S (OMX: GEN) gab heute bekannt, dass das Unternehmen nach den Bedingungen der Kooperation mit GlaxoSmithKline (GSK) den sechsten Entwicklungsmeilenstein für Ofatumumab (HuMax-CD20(R)) erreicht hat. Durch die Behandlung des ersten Patienten im Rahmen der Studie der Phase I mit Ofatumumab bei rezidivierendem bzw. refraktärem follikulärem Non-Hodgkin-Lymphom und chronischer lymphatischer Leukämie in Japan, wurde eine Meilensteinzahlung in Höhe von ca. 29 Mio. DKK (ca. 5,6 Mio. USD) ausgelöst.
Bei Ofatumumab handelt es sich um einen in der Untersuchung befindlichen, humanen monoklonalen Antikörper einer neuen Generation, der an eine spezifische, membranproximale kleine Epitop-Schleife (spezifische Bindungsstelle) des CD20-Moleküls auf der Oberfläche von B-Zellen bindet. Ofatumumab wird im Rahmen eines gemeinsamen Entwicklungs- und Vertriebsabkommens von GlaxoSmithKline und Genmab gemeinsam zur Behandlung der chronischen lymphatischen Leukämie, des follikulären Non-Hodgkin-Lymphoms , des diffusen grosszelligen B-Zellen-Lymphoms, der rheumatoiden Arthritis und der schubförmig remittierenden Multiplen Sklerose entwickelt. Der Wirkstoff ist bisher noch in keinem Land zum Verkauf zugelassen.
mfg ipollit
Genmab erreicht Meilenstein bei der Ofatumumab-Kooperation
KOPENHAGEN, Dänemark, October 10 (ots/PRNewswire) - -
Zusammenfassung: Genmab erreicht sechsten Meilenstein in der Ofatumumab-Kooperation mit GSK
Genmab A/S (OMX: GEN) gab heute bekannt, dass das Unternehmen nach den Bedingungen der Kooperation mit GlaxoSmithKline (GSK) den sechsten Entwicklungsmeilenstein für Ofatumumab (HuMax-CD20(R)) erreicht hat. Durch die Behandlung des ersten Patienten im Rahmen der Studie der Phase I mit Ofatumumab bei rezidivierendem bzw. refraktärem follikulärem Non-Hodgkin-Lymphom und chronischer lymphatischer Leukämie in Japan, wurde eine Meilensteinzahlung in Höhe von ca. 29 Mio. DKK (ca. 5,6 Mio. USD) ausgelöst.
Bei Ofatumumab handelt es sich um einen in der Untersuchung befindlichen, humanen monoklonalen Antikörper einer neuen Generation, der an eine spezifische, membranproximale kleine Epitop-Schleife (spezifische Bindungsstelle) des CD20-Moleküls auf der Oberfläche von B-Zellen bindet. Ofatumumab wird im Rahmen eines gemeinsamen Entwicklungs- und Vertriebsabkommens von GlaxoSmithKline und Genmab gemeinsam zur Behandlung der chronischen lymphatischen Leukämie, des follikulären Non-Hodgkin-Lymphoms , des diffusen grosszelligen B-Zellen-Lymphoms, der rheumatoiden Arthritis und der schubförmig remittierenden Multiplen Sklerose entwickelt. Der Wirkstoff ist bisher noch in keinem Land zum Verkauf zugelassen.
mfg ipollit
OFATUMUMAB INDUCES LONG LASTING CLINICAL RESPONSES IN PHASE II RA STUDY
Data to be presented at ACR meeting
Copenhagen, Denmark; October 8, 2008 – Genmab A/S (OMX: GEN) announced today that data showing that rheumatoid arthritis patients who participated in the ofatumumab (HuMax-CD20®) Phase II study achieved long lasting results at the 48 week follow up period. The data will be presented in a poster session at the ACR/ARHP 2008 Annual Scientific Meeting in San Francisco, California on October 26, 2008. The full abstract can be found by clicking this link, then clicking http://www.abstractsonline.com/plan/start.aspx?mkey={5880E48… Advanced Search and typing "ofatumumab" in the Abstract Body field and pressing Enter.
Ofatumumab is an investigational, new generation, human monoclonal antibody that targets a distinct membrane proximal, small loop epitope (specific binding site) of the CD20 molecule on the surface of B-cells. Ofatumumab is being developed to treat chronic lymphocytic leukemia, follicular non-Hodgkin's lymphoma, diffuse large B-cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis under a co-development and commercialization agreement between Genmab and GlaxoSmithKline. It is not yet approved for sale in any country.
mfg ipollit
Data to be presented at ACR meeting
Copenhagen, Denmark; October 8, 2008 – Genmab A/S (OMX: GEN) announced today that data showing that rheumatoid arthritis patients who participated in the ofatumumab (HuMax-CD20®) Phase II study achieved long lasting results at the 48 week follow up period. The data will be presented in a poster session at the ACR/ARHP 2008 Annual Scientific Meeting in San Francisco, California on October 26, 2008. The full abstract can be found by clicking this link, then clicking http://www.abstractsonline.com/plan/start.aspx?mkey={5880E48… Advanced Search and typing "ofatumumab" in the Abstract Body field and pressing Enter.
Ofatumumab is an investigational, new generation, human monoclonal antibody that targets a distinct membrane proximal, small loop epitope (specific binding site) of the CD20 molecule on the surface of B-cells. Ofatumumab is being developed to treat chronic lymphocytic leukemia, follicular non-Hodgkin's lymphoma, diffuse large B-cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis under a co-development and commercialization agreement between Genmab and GlaxoSmithKline. It is not yet approved for sale in any country.
mfg ipollit
Rituxan... der Konkurrent von Genmab's Ofatumumab gegen CLL. Ofatumumab richtet sich wie Rituxan gegen CD20, soll aber erheblich besser wirken bei CLL als Rituxan.
Phase III Study of Rituxan in Leukemia Meets its Goal
By Catherine Hollingsworth
Staff Writer
Genentech Inc. and Biogen Idec Inc. said their investigational leukemia drug Rituxan (rituximab) passed a second Phase III study, meeting its primary goal of improving the period of time before the disease progresses compared to chemotherapy alone.
Rituxan combined with chemotherapy treatments fludarabine and cyclophosphamide improved progression-free survival in patients with previously treated CD20-positive chronic lymphocytic leukemia (CLL).
Earlier this year, another European Phase III study, CLL-8, showed a similar treatment combination improved PFS in patients with CLL who previously had not received treatment.
South San Francisco-based Genentech still is reviewing the data, the details of which will be presented at a future medical meeting.
An independent review of the primary endpoint is being conducted for U.S. regulatory purposes.
The study, known as REACH (Rituximab in thE study of relApsed Chronic lympHocytic leukemia), is the largest ever in relapsed CLL. It is the first such study to show an improvement in progression-free survival, according to Genentech.
Rituxan, currently approved for rheumatoid arthritis and non-Hodgkin's lymphoma, has been associated with a life-threatening brain infection known as progressive multifocal leukoencephalopathy, or PML. But there were no new or unexpected safety signals reported in the leukemia study, Genentech said.
Genentech and Biogen Idec co-market Rituxan in the U.S., and Roche markets it as MabThera in the rest of the world, except Japan, where Rituxan is co-marketed by Chugai and Zenyaku Kogyo Co. Ltd.
Cephalon Inc's chemotherapy agent Treanda (bendamustine HCl) is among the current treatments approved for CLL. That approval was based on a 301-patient study showing a higher overall response compared to standard chemo agent chlorambucil. That study also showed significantly longer progression-free survival (18 months vs. six months) and a longer-lasting response compared with another chemo agent chlorambucil (19 moths vs. seven months). (See BioWorld Today, March 21, 2008.)
A Cephalon-sponsored Phase II study will evaluate Treanda and Rituxan in elderly patients with CLL. Genentech is not involved with that trial.
Other currently available treatments for CLL include the monoclonal antibody Campath (alemtuzumab), which targets the CD52 antigen, and chemotherapy agents fludarabine and cyclophosphamide, according to the American Cancer Society. Known as the FR regimen, those two chemo drugs were studied alongside Rituxan in the Genentech-Biogen Idec Phase III study of relapsed leukemia patients.
The FR regimen also has been studied alongside another potential antibody for CLL: Genta Inc.'s DNA-based drug Genasense.
Berkeley Height, N.J.-based Genta filed an amended new drug application for Genasense following an appeal of a December 2006 approvable letter. FDA's action date on that resubmission is set for Dec. 3. (See BioWorld Today, July 15, 2008.)
The major goal of the Phase III Genasense study was to put relapsing leukemia patients into complete remission - and that endpoint was "very solid," Raymond Warrell, CEO of Genta, told BioWorld Today.
Combining Genasense with the FR chemotherapy regimen significantly increased the complete remission rate, he said, with a median duration of almost two years longer than those on chemo.
Warrell contrasted the remission endpoint in the Genasense study with the progression-free survival (PFS) endpoint used in the Rituxan study. With PFS as the objective, he said, patients don't necessarily have to respond or show a benefit but must show a delay in the regrowth of the disease.
Danish biotech GenMab A/S and partner GlaxoSmithKline plc have a leukemia drug directed at the same target as Rituxan's - the CD20 molecule on B cells. That product also met its endpoint in a Phase III pivotal study. (See BioWorld Today, Aug. 1, 2008.)
Richard Parks, a senior analyst with Piper Jaffray, wrote in a research note that "ofatumumab has shown compelling activity as a single agent."
There is a significant amount of Rituxan use as a single agent for CLL, he said, "despite limited evidence of activity" because many patients cannot tolerate myelosuppressive chemotherapy.
Geoffrey Nichol, senior vice president of product development at Medarex Inc., which has a Phase I antibody in development for CLL, told BioWorld Today that his company's product, which binds to CD19, may be expressed in more cells and in more patients than CD20, offering a possible advantage.
Shares in Genentech (NYSE: DNA) were down $2.43, closing at $78.19.
Published October 8, 2008
mfg ipollit
Phase III Study of Rituxan in Leukemia Meets its Goal
By Catherine Hollingsworth
Staff Writer
Genentech Inc. and Biogen Idec Inc. said their investigational leukemia drug Rituxan (rituximab) passed a second Phase III study, meeting its primary goal of improving the period of time before the disease progresses compared to chemotherapy alone.
Rituxan combined with chemotherapy treatments fludarabine and cyclophosphamide improved progression-free survival in patients with previously treated CD20-positive chronic lymphocytic leukemia (CLL).
Earlier this year, another European Phase III study, CLL-8, showed a similar treatment combination improved PFS in patients with CLL who previously had not received treatment.
South San Francisco-based Genentech still is reviewing the data, the details of which will be presented at a future medical meeting.
An independent review of the primary endpoint is being conducted for U.S. regulatory purposes.
The study, known as REACH (Rituximab in thE study of relApsed Chronic lympHocytic leukemia), is the largest ever in relapsed CLL. It is the first such study to show an improvement in progression-free survival, according to Genentech.
Rituxan, currently approved for rheumatoid arthritis and non-Hodgkin's lymphoma, has been associated with a life-threatening brain infection known as progressive multifocal leukoencephalopathy, or PML. But there were no new or unexpected safety signals reported in the leukemia study, Genentech said.
Genentech and Biogen Idec co-market Rituxan in the U.S., and Roche markets it as MabThera in the rest of the world, except Japan, where Rituxan is co-marketed by Chugai and Zenyaku Kogyo Co. Ltd.
Cephalon Inc's chemotherapy agent Treanda (bendamustine HCl) is among the current treatments approved for CLL. That approval was based on a 301-patient study showing a higher overall response compared to standard chemo agent chlorambucil. That study also showed significantly longer progression-free survival (18 months vs. six months) and a longer-lasting response compared with another chemo agent chlorambucil (19 moths vs. seven months). (See BioWorld Today, March 21, 2008.)
A Cephalon-sponsored Phase II study will evaluate Treanda and Rituxan in elderly patients with CLL. Genentech is not involved with that trial.
Other currently available treatments for CLL include the monoclonal antibody Campath (alemtuzumab), which targets the CD52 antigen, and chemotherapy agents fludarabine and cyclophosphamide, according to the American Cancer Society. Known as the FR regimen, those two chemo drugs were studied alongside Rituxan in the Genentech-Biogen Idec Phase III study of relapsed leukemia patients.
The FR regimen also has been studied alongside another potential antibody for CLL: Genta Inc.'s DNA-based drug Genasense.
Berkeley Height, N.J.-based Genta filed an amended new drug application for Genasense following an appeal of a December 2006 approvable letter. FDA's action date on that resubmission is set for Dec. 3. (See BioWorld Today, July 15, 2008.)
The major goal of the Phase III Genasense study was to put relapsing leukemia patients into complete remission - and that endpoint was "very solid," Raymond Warrell, CEO of Genta, told BioWorld Today.
Combining Genasense with the FR chemotherapy regimen significantly increased the complete remission rate, he said, with a median duration of almost two years longer than those on chemo.
Warrell contrasted the remission endpoint in the Genasense study with the progression-free survival (PFS) endpoint used in the Rituxan study. With PFS as the objective, he said, patients don't necessarily have to respond or show a benefit but must show a delay in the regrowth of the disease.
Danish biotech GenMab A/S and partner GlaxoSmithKline plc have a leukemia drug directed at the same target as Rituxan's - the CD20 molecule on B cells. That product also met its endpoint in a Phase III pivotal study. (See BioWorld Today, Aug. 1, 2008.)
Richard Parks, a senior analyst with Piper Jaffray, wrote in a research note that "ofatumumab has shown compelling activity as a single agent."
There is a significant amount of Rituxan use as a single agent for CLL, he said, "despite limited evidence of activity" because many patients cannot tolerate myelosuppressive chemotherapy.
Geoffrey Nichol, senior vice president of product development at Medarex Inc., which has a Phase I antibody in development for CLL, told BioWorld Today that his company's product, which binds to CD19, may be expressed in more cells and in more patients than CD20, offering a possible advantage.
Shares in Genentech (NYSE: DNA) were down $2.43, closing at $78.19.
Published October 8, 2008
mfg ipollit
Genmab...
Danish rumour brings Glaxo into the spotlight
Pharmaceuticals group GlaxoSmithKline was in the spotlight amid speculation that it is in talks to buy Danish biotechnology company Genmab.
By Ben Harrington
Last Updated: 10:05PM BST 03 Oct 2008
In 2006, GSK acquired the rights for a leukaemia drug developed by Genmab and now has a 9.9pc stake in the company, which is valued at Dkr 13.1bn (£1.3bn).
According to sources, several months ago Genmab hired investment bank Merrill Lynch to advise on transaction and held talks with potential buyers.
US-based Biogen Idec and Denmark's Novo Nordisk have both been tipped as potential buyers in the past.
There was also talk that US activist investor Carl Icahn, who is chairman of Imclone, may be interested in buying a stake.
Informed sources poured cold water on the GSK takeover tale and the pharmaceutical heavyweight is not thought to be in talks to acquire Genmab.
Nevertheless, traders still kept their eye on Genmab as there is a possibility of deal in the future. GSK shares perked up 4 to £12.29.
mfg ipollit
Danish rumour brings Glaxo into the spotlight
Pharmaceuticals group GlaxoSmithKline was in the spotlight amid speculation that it is in talks to buy Danish biotechnology company Genmab.
By Ben Harrington
Last Updated: 10:05PM BST 03 Oct 2008
In 2006, GSK acquired the rights for a leukaemia drug developed by Genmab and now has a 9.9pc stake in the company, which is valued at Dkr 13.1bn (£1.3bn).
According to sources, several months ago Genmab hired investment bank Merrill Lynch to advise on transaction and held talks with potential buyers.
US-based Biogen Idec and Denmark's Novo Nordisk have both been tipped as potential buyers in the past.
There was also talk that US activist investor Carl Icahn, who is chairman of Imclone, may be interested in buying a stake.
Informed sources poured cold water on the GSK takeover tale and the pharmaceutical heavyweight is not thought to be in talks to acquire Genmab.
Nevertheless, traders still kept their eye on Genmab as there is a possibility of deal in the future. GSK shares perked up 4 to £12.29.
mfg ipollit
Viropharma...
(das im Text unten genannte Szenario 1 ist inzwischen in etwa eingetreten...)
http://seekingalpha.com/article/99197-viropharma-lev-pharmac…
Viropharma, Lev Pharmaceuticals: An Attractive Biotech Pair
by: Dan Weiss posted on: October 09, 2008 | about stocks: LEVP.OB / VPHM
Lev Pharmaceuticals (LEVP.OB) is a biotechnology company, which has focused on developing a treatment for the serious, and potentially life-threatening disorder known as hereditary angioedema, which has no FDA approved treatments in the United States. Lev's treatment is known as Cinryze and the company is seeking approval for both acute and prophylactic indications with a PDUFA date of October 14, 2008.
In late January 2008, Lev received a complete response letter from the FDA for the acute indication seeking additional information regarding manufacturing, chemistry and controls used in the studies. Lev did not present the prophylactic treatment for approval for the late January 2008 decision. In early May, Lev presented in-depth findings to an FDA advisory panel for the prophylactic treatment of HAE where the panel unanimously recommended approval to the FDA which will be making its decision next week.
The action by the FDA advisory panel significantly increases the odds of approval by the FDA and the safety record of similar treatments in Europe has been excellent. I have spoken with several physicians who speak very highly of the treatment and feel that it will have significant demand right out of the gate and that the studies show that the treatment is safe and greatly improves the quality of life with those affected by HAE.
On July 15, 2008, Viropharma (VPHM) announced that they would be acquiring Lev Pharmaceuticals in a cash and stock deal worth approximately $443 million ($2.75/share) in upfront considerations with the potential for an additional $174.6 million ($1.00/share) in cash payments to Lev shareholders if certain milestones are met. The upfront considerations consist of $2.25 in cash plus $0.50 in shares of Viropharma (subject to a collar ranging from $10.03-15.68). Additional potential milestones include up to $1.00 per share in cash to shareholders of Lev under the following 2 circumstances: 1) $0.50 per share consideration under 2 potential scenarios: a) Cinryze is approved for acute treatment of HAE and orphan exclusivity is granted or b) orphan exclusivity for the acute treatment of HAE has not become effective for anyone for two years from the date of closing and date of FDA approval for prophylaxis, whichever is later; and 2) an additional $0.50 per share consideration when Cinryze reaches $600 million in cumulative net sales within 10 years of closing.
The deal was approved by both boards in July and was later approved by the FTC on September 3, 2008. As mentioned earlier, the PDUFA date from the FDA for both the acute and prophylactic indication is October 14, 2008. Following the FDA decision, shareholders of Lev will vote on the merger on October 21, 2008 with the closing of the deal occurring on October 23, 2008. In terms of what could derail the merger, the only major potential stumbling block would be if the FDA were to send Lev non-approvable letters for both the prophylactic and acute indications, which is very highly unlikely.
Potential Outcomes
1) FDA approval for prophylactic with the request for additional information on acute: This in my opinion is the most likely outcome and would be positive for both shareholders of Lev and Viropharma as Cinryze would be able to move on to the market very quickly (remember there is no other approved treatments in the U.S. at this time) and significantly improve the lives of those inflicted with HAE. Viropharma would benefit from nearly immediate revenues and cash flows flowing to the company and allowing for the generation of additional product lines for the company to improve overall visibility on revenues and profits. In this scenario, Viropharma would be able to request approval for acute once any questions are addressed and it would still be likely that Lev would be the first to market on the acute side. I would note that no other companies are currently attempting to gain approval on the prophylactic side for HAE.
2) FDA approval for both prophylactic and acute indications. This would of course be the absolute best-case scenario for both Lev and Viropharma. Under this scenario, Lev shareholders would get the full $2.75 in upfront payments plus it is highly likely that they would received the additional $1.00 in milestone payments. Viropharma would of course greatly benefit from this scenario as well since market exclusivity would likely be granted for Cinryze for a period of 7 years thus very much increasing the likelihood of very strong profitability and cash flows for the company for several years to come.
3) The FDA requests additional information for both indications. If it is a complete response letter with simple to address issues then the merger will likely still occur but in the unlikely event that additional clinical studies are required for both acute and prophylactic then Viropharma could decide to exit the deal. I have been in close contact with several people both within Lev and physicians and it would be extremely unlikely especially after the FDA advisory panel's opinion in May for an outcome to occur which would endanger the merger.
If Lev were to gain approval for Cinryze from the FDA they would be likely be given market exclusivity for a period of seven years. The estimated potential market for Cinryze is conservatively estimated at $200-300 million per year during the exclusivity period with an additional potential catalyst being any approval in later years for the treatment of heart attacks, which is also being studied by the company.
Valuation Targets
Clearly, the target for Lev Pharmaceuticals is the $2.75 acquisition price under the most likely scenario. There is the potential for up to $1.00 in additional value after the deal closes to as long as ten years out. It is very likely in my opinion that the second 0.50 will trigger within the 10 year period as Cinryze sales (assuming approval) should easily exceed $600 million on a cumulative basis. Discounting the two potential 0.50 payments gives a fair value of approximately $3 or slightly higher per share.
For Viropharma, the company will be using almost all cash for this deal with only a small number of shares that will need to be added to the float. In addition, the company has an additional product in the pipeline and will have strong cash flows especially while Cinryze and Vancocin overlap. Cinryze also has the longer term potential to help with heart attacks which if eventually approved would add significant revenues to the company several years down the road. Based strictly on the approval of Cinryze and the continued use of Vancocin and the healthy balance sheet of the company, I value the shares at approximately $20/share.
Disclosure: Author is long Lev Pharmaceuticals and Viropharma
mfg ipollit
(das im Text unten genannte Szenario 1 ist inzwischen in etwa eingetreten...)
http://seekingalpha.com/article/99197-viropharma-lev-pharmac…
Viropharma, Lev Pharmaceuticals: An Attractive Biotech Pair
by: Dan Weiss posted on: October 09, 2008 | about stocks: LEVP.OB / VPHM
Lev Pharmaceuticals (LEVP.OB) is a biotechnology company, which has focused on developing a treatment for the serious, and potentially life-threatening disorder known as hereditary angioedema, which has no FDA approved treatments in the United States. Lev's treatment is known as Cinryze and the company is seeking approval for both acute and prophylactic indications with a PDUFA date of October 14, 2008.
In late January 2008, Lev received a complete response letter from the FDA for the acute indication seeking additional information regarding manufacturing, chemistry and controls used in the studies. Lev did not present the prophylactic treatment for approval for the late January 2008 decision. In early May, Lev presented in-depth findings to an FDA advisory panel for the prophylactic treatment of HAE where the panel unanimously recommended approval to the FDA which will be making its decision next week.
The action by the FDA advisory panel significantly increases the odds of approval by the FDA and the safety record of similar treatments in Europe has been excellent. I have spoken with several physicians who speak very highly of the treatment and feel that it will have significant demand right out of the gate and that the studies show that the treatment is safe and greatly improves the quality of life with those affected by HAE.
On July 15, 2008, Viropharma (VPHM) announced that they would be acquiring Lev Pharmaceuticals in a cash and stock deal worth approximately $443 million ($2.75/share) in upfront considerations with the potential for an additional $174.6 million ($1.00/share) in cash payments to Lev shareholders if certain milestones are met. The upfront considerations consist of $2.25 in cash plus $0.50 in shares of Viropharma (subject to a collar ranging from $10.03-15.68). Additional potential milestones include up to $1.00 per share in cash to shareholders of Lev under the following 2 circumstances: 1) $0.50 per share consideration under 2 potential scenarios: a) Cinryze is approved for acute treatment of HAE and orphan exclusivity is granted or b) orphan exclusivity for the acute treatment of HAE has not become effective for anyone for two years from the date of closing and date of FDA approval for prophylaxis, whichever is later; and 2) an additional $0.50 per share consideration when Cinryze reaches $600 million in cumulative net sales within 10 years of closing.
The deal was approved by both boards in July and was later approved by the FTC on September 3, 2008. As mentioned earlier, the PDUFA date from the FDA for both the acute and prophylactic indication is October 14, 2008. Following the FDA decision, shareholders of Lev will vote on the merger on October 21, 2008 with the closing of the deal occurring on October 23, 2008. In terms of what could derail the merger, the only major potential stumbling block would be if the FDA were to send Lev non-approvable letters for both the prophylactic and acute indications, which is very highly unlikely.
Potential Outcomes
1) FDA approval for prophylactic with the request for additional information on acute: This in my opinion is the most likely outcome and would be positive for both shareholders of Lev and Viropharma as Cinryze would be able to move on to the market very quickly (remember there is no other approved treatments in the U.S. at this time) and significantly improve the lives of those inflicted with HAE. Viropharma would benefit from nearly immediate revenues and cash flows flowing to the company and allowing for the generation of additional product lines for the company to improve overall visibility on revenues and profits. In this scenario, Viropharma would be able to request approval for acute once any questions are addressed and it would still be likely that Lev would be the first to market on the acute side. I would note that no other companies are currently attempting to gain approval on the prophylactic side for HAE.
2) FDA approval for both prophylactic and acute indications. This would of course be the absolute best-case scenario for both Lev and Viropharma. Under this scenario, Lev shareholders would get the full $2.75 in upfront payments plus it is highly likely that they would received the additional $1.00 in milestone payments. Viropharma would of course greatly benefit from this scenario as well since market exclusivity would likely be granted for Cinryze for a period of 7 years thus very much increasing the likelihood of very strong profitability and cash flows for the company for several years to come.
3) The FDA requests additional information for both indications. If it is a complete response letter with simple to address issues then the merger will likely still occur but in the unlikely event that additional clinical studies are required for both acute and prophylactic then Viropharma could decide to exit the deal. I have been in close contact with several people both within Lev and physicians and it would be extremely unlikely especially after the FDA advisory panel's opinion in May for an outcome to occur which would endanger the merger.
If Lev were to gain approval for Cinryze from the FDA they would be likely be given market exclusivity for a period of seven years. The estimated potential market for Cinryze is conservatively estimated at $200-300 million per year during the exclusivity period with an additional potential catalyst being any approval in later years for the treatment of heart attacks, which is also being studied by the company.
Valuation Targets
Clearly, the target for Lev Pharmaceuticals is the $2.75 acquisition price under the most likely scenario. There is the potential for up to $1.00 in additional value after the deal closes to as long as ten years out. It is very likely in my opinion that the second 0.50 will trigger within the 10 year period as Cinryze sales (assuming approval) should easily exceed $600 million on a cumulative basis. Discounting the two potential 0.50 payments gives a fair value of approximately $3 or slightly higher per share.
For Viropharma, the company will be using almost all cash for this deal with only a small number of shares that will need to be added to the float. In addition, the company has an additional product in the pipeline and will have strong cash flows especially while Cinryze and Vancocin overlap. Cinryze also has the longer term potential to help with heart attacks which if eventually approved would add significant revenues to the company several years down the road. Based strictly on the approval of Cinryze and the continued use of Vancocin and the healthy balance sheet of the company, I value the shares at approximately $20/share.
Disclosure: Author is long Lev Pharmaceuticals and Viropharma
mfg ipollit
Antwort auf Beitrag Nr.: 35.532.124 von ipollit am 11.10.08 17:05:34VPHM... die FDA hat wie erwartet Cinryze als Prophylaxe gegen HAE zugelassen... damit ist es das erste Medikament dieser Art in den USA. Der Antrag von Cinryze als akute Behandlung von HAE wurde auf Anraten der FDA zuvor zurückgezogen.
UPDATE 1-US clears Lev Pharma drug for rare swelling disease
Fri Oct 10, 2008 5:56pm EDT
(Adds FDA and company comment, background)
WASHINGTON, Oct 10 (Reuters) - U.S. health regulators have approved Lev Pharmaceuticals Inc's (LEVP.OB: Quote, Profile, Research, Stock Buzz) experimental drug to treat a rare genetic disease that causes swelling of the hands, feet and other parts of the body.
The drug, called Cinryze, aims to treat hereditary angioedema, a spontaneous and potentially deadly disorder that can be triggered by stress, surgery or infection, the U.S. Food and Drug Administration said in a statement on Friday.
ViroPharma Inc (VPHM.O: Quote, Profile, Research, Stock Buzz) has agreed to buy Lev in a $443 million deal expected to be completed by the end of the year.
Hereditary angioedema disease also can cause swelling of the airway and larynx, which can make it impossible for those experiencing it to breathe and cause asphyxiation, the FDA said.
Up to 10,000 people in the United States have the condition, according to the agency.
Lev's intravenous drug, a protein made from human plasma, is given every three or four days to help prevent swelling, it added.
"Cinryze should greatly enhance treatment options for those with hereditary angioedema and potentially save lives," Dr. Jesse Goodman, head of the FDA's Center for Biologics Evaluation and Research, said.
The drug should be available for patients later this year, Lev said in a statement.
Company studies show Cinryze reduces the frequency of attacks or prevents them in most patients, the company said. Common side effects include rash near the injection site and lightheadedness.
The drugmaker is working to ensure that health insurers cover the product, it added.
Before the FDA announcement, shares of Lev rose more than 8 percent to close at $2.38 while shares of ViroPharma closed up 0.8 percent at $9.57. (Reporting by Susan Heavey; Editing by Tim Dobbyn and Carol Bishopric)
************
Based on discussions with FDA, Lev has requested the withdrawal of the portion of the application referring to data for the acute treatment of HAE attacks. This data will be resubmitted as a supplemental BLA, along with additional data from ongoing open label acute studies of Cinryze, as soon as possible. The company does not believe that an additional study will be required, but FDA must agree.
mfg ipollit
UPDATE 1-US clears Lev Pharma drug for rare swelling disease
Fri Oct 10, 2008 5:56pm EDT
(Adds FDA and company comment, background)
WASHINGTON, Oct 10 (Reuters) - U.S. health regulators have approved Lev Pharmaceuticals Inc's (LEVP.OB: Quote, Profile, Research, Stock Buzz) experimental drug to treat a rare genetic disease that causes swelling of the hands, feet and other parts of the body.
The drug, called Cinryze, aims to treat hereditary angioedema, a spontaneous and potentially deadly disorder that can be triggered by stress, surgery or infection, the U.S. Food and Drug Administration said in a statement on Friday.
ViroPharma Inc (VPHM.O: Quote, Profile, Research, Stock Buzz) has agreed to buy Lev in a $443 million deal expected to be completed by the end of the year.
Hereditary angioedema disease also can cause swelling of the airway and larynx, which can make it impossible for those experiencing it to breathe and cause asphyxiation, the FDA said.
Up to 10,000 people in the United States have the condition, according to the agency.
Lev's intravenous drug, a protein made from human plasma, is given every three or four days to help prevent swelling, it added.
"Cinryze should greatly enhance treatment options for those with hereditary angioedema and potentially save lives," Dr. Jesse Goodman, head of the FDA's Center for Biologics Evaluation and Research, said.
The drug should be available for patients later this year, Lev said in a statement.
Company studies show Cinryze reduces the frequency of attacks or prevents them in most patients, the company said. Common side effects include rash near the injection site and lightheadedness.
The drugmaker is working to ensure that health insurers cover the product, it added.
Before the FDA announcement, shares of Lev rose more than 8 percent to close at $2.38 while shares of ViroPharma closed up 0.8 percent at $9.57. (Reporting by Susan Heavey; Editing by Tim Dobbyn and Carol Bishopric)
************
Based on discussions with FDA, Lev has requested the withdrawal of the portion of the application referring to data for the acute treatment of HAE attacks. This data will be resubmitted as a supplemental BLA, along with additional data from ongoing open label acute studies of Cinryze, as soon as possible. The company does not believe that an additional study will be required, but FDA must agree.
mfg ipollit
Hallo ipollit,
ich hoffe dein Depot hat es nicht zu sehr zerissen in den letzten Tagen. Mich interessiert natürlich von deinen Werten hauptsächlich Evotec. Hast du reduziert, gehalten oder verbilligt. Mit dem P2X7-Rezeptorantagonisten sind sie letzte Woche in die erste Phase gegangen, ist leider in dem allgemeinen Chaos total untergegangen.
Was hälst du davon?
Important news flow in 08 and 09
Partnering of EVT 201 (2008)
Phase II proof-of-concept for EVT 302 (2009)
Start of Phase II with EVT 101 (2008)
Phase II proof-of-concept for EVT 101 (End of 2009 / Beginning of 2010)
Phase I results for VR1 and P2X7(2009)
Start of Phase I with P2X3 (2009)
Potential further partnerships
Development and commercialization partnerships for clinical assets
Preclinical discovery alliances
Quelle: http://www.evotec.com/en/pdf/investors/Evotec_10-2008.pdf
Ich finde, so etwas macht man nicht in der Form public, wenn man nicht schon sehr weit mit den Verhandlungen wäre. Vor einigen Monaten sprach man ja noch davon, dass es der Markt noch in 2008 erwartet!
Außerdem gibt es ein Interview, in dem er angeblich von... There are some important meetings coming up next 2 to 6 weeks.... spricht. Das ist jetzt ca. 2-3 Wochen her. Also für mich verdichten sich die Zeichen, nur der Kurs sieht es anders.
Wäre schön, wenn du wieder etwas Zeit findest, um auf dem Evotec Board deine Meinung abzugeben, selbst zenman "ahnt" jetzt schon einen 3€ Hype!
Grüße und VIEL GLÜCK in diesen wilden Zeiten!!!
ich hoffe dein Depot hat es nicht zu sehr zerissen in den letzten Tagen. Mich interessiert natürlich von deinen Werten hauptsächlich Evotec. Hast du reduziert, gehalten oder verbilligt. Mit dem P2X7-Rezeptorantagonisten sind sie letzte Woche in die erste Phase gegangen, ist leider in dem allgemeinen Chaos total untergegangen.
Was hälst du davon?
Important news flow in 08 and 09
Partnering of EVT 201 (2008)
Phase II proof-of-concept for EVT 302 (2009)
Start of Phase II with EVT 101 (2008)
Phase II proof-of-concept for EVT 101 (End of 2009 / Beginning of 2010)
Phase I results for VR1 and P2X7(2009)
Start of Phase I with P2X3 (2009)
Potential further partnerships
Development and commercialization partnerships for clinical assets
Preclinical discovery alliances
Quelle: http://www.evotec.com/en/pdf/investors/Evotec_10-2008.pdf
Ich finde, so etwas macht man nicht in der Form public, wenn man nicht schon sehr weit mit den Verhandlungen wäre. Vor einigen Monaten sprach man ja noch davon, dass es der Markt noch in 2008 erwartet!
Außerdem gibt es ein Interview, in dem er angeblich von... There are some important meetings coming up next 2 to 6 weeks.... spricht. Das ist jetzt ca. 2-3 Wochen her. Also für mich verdichten sich die Zeichen, nur der Kurs sieht es anders.
Wäre schön, wenn du wieder etwas Zeit findest, um auf dem Evotec Board deine Meinung abzugeben, selbst zenman "ahnt" jetzt schon einen 3€ Hype!
Grüße und VIEL GLÜCK in diesen wilden Zeiten!!!
Antwort auf Beitrag Nr.: 35.546.581 von orfmen am 13.10.08 12:10:46hallo orfmen
für eine (technische) Reduzierung war es zu spät, zukaufen will ich jetzt auch nicht, da Evotec zwar einigermaßen breit aufgestellt ist und ich die MK für akzeptabel halte, aber das Risiko doch vorhanden ist, dass sich eine Menge Flops in der Pipeline befinden.
Ich würde nicht darauf setzten, dass ein EVT-201 auf jeden Fall noch in 2008 zustande kommt. Es wird schon deutlich, dass es schwierig ist, einen Partner zu finden. Dass mit den "wichtigen Gesprächen in den nächsten 2 bis 6 Wochen" wurde von Aldag auf der letzten UBS-Konferenz gesagt. Mal sehen, ob sich daraus etwas ergibt.
mfg ipollit
für eine (technische) Reduzierung war es zu spät, zukaufen will ich jetzt auch nicht, da Evotec zwar einigermaßen breit aufgestellt ist und ich die MK für akzeptabel halte, aber das Risiko doch vorhanden ist, dass sich eine Menge Flops in der Pipeline befinden.
Ich würde nicht darauf setzten, dass ein EVT-201 auf jeden Fall noch in 2008 zustande kommt. Es wird schon deutlich, dass es schwierig ist, einen Partner zu finden. Dass mit den "wichtigen Gesprächen in den nächsten 2 bis 6 Wochen" wurde von Aldag auf der letzten UBS-Konferenz gesagt. Mal sehen, ob sich daraus etwas ergibt.
mfg ipollit
Genmab's HuMax-CD20 gegen RA...
http://www.nature.com/nbt/journal/v26/n10/full/nbt1008-1053.…
Nature Biotechnology 26, 1053 - 1054 (2008)
doi:10.1038/nbt1008-1053
CD20 blockers eye crowded rheumatology market
George S Mack1
Columbia, South Carolina
When trial results for a novel cancer drug were trumpeted in July, the rheumatology field felt the ripples. The drug is ofatumumab, a monoclonal antibody (mAb) targeting the CD20 molecule on B lymphocytes. Its makers, London-based GlaxoSmithKline (GSK) and Genmab of Copenhagen, announced that the mAb had met its primary and secondary endpoints in chronic lymphocytic leukemia (CLL). But with two double-blind phase 3 studies of ofatumumab for rheumatoid arthritis already underway in Italy and Eastern Europe, these companies are clearly looking beyond cancer indications to lucrative autoimmune disease markets for their second-generation molecule.
The idea is to follow the trail blazed by Genentech of South San Francisco, Roche of Basel and Biogen-Idec of Cambridge, Massachusetts, with Rituxan (rituximab), their anti-CD20 chimeric mAb. Rituxan was approved by the US Food and Drug Administration to treat B-cell lymphomas and leukemias in 1997 and then as a third-line therapy against rheumatoid arthritis in 2006. Currently marketed by Roche in Europe as MabThera, it surpassed $5 billion in worldwide sales in 2007 alone.
GSK has invested heavily in ofatumumab. The company made headlines at the end of 2006 when the licensing agreement to co-develop this fully human IgG1- mAb for cancer and rheumatoid arthritis was announced. The deal, potentially worth $2.1 billion, saw Genmab receive an upfront $102 million as a sweetener for their anti-CD20 mAb. GSK also took a 10% stake in the biotech firm by purchasing $357 million in shares. The transaction was deemed to be the largest ever in Europe for a single product; if all milestones are met it will exceed the $2.0 billion deal between New York–based Bristol-Myers Squibb and New York–based ImClone Systems for blockbuster cancer chimeric mAb Erbitux (cetuximab).
The meteoric rise of tumor necrosis factor (TNF)- blockers over the past decade ushered in the age of biologic therapies for rheumatoid arthritis. Anti-TNF agents—Enbrel (etanercept), Humira (adalimumab) and Remicade (infliximab)—are now labeled for second-line use after failure of methotrexate and nonsteroidal anti-inflammatory drugs (NSAIDs). But there is no product that works for everyone—about a third of those with rheumatoid arthritis do not respond to first- or second-line therapy.
Rituxan is currently a third-line therapy for rheumatoid arthritis after failure of methotrexate and anti-TNF- products. "We'd like to try it in patients much earlier on, and we're working towards that at the moment," says immunologist Geraldine Cambridge, who has collaborated for many years with rheumatologist Jonathan Edwards, both of the Center for Rheumatology Research at University College London. The two have done much of the basic science groundwork as well as overseen clinical trials over the past decade to establish the effectiveness of B-cell depletion with CD20 blockade. "You need to get these patients in remission as early as possible," she says. "If you lose one year while patients are slowly failing on methotrexate and TNF-inhibitors, that means they've lost their job by then and will probably never get back to work again."
The clinical approach by which B cells are ablated to treat rheumatoid arthritis has only recently gained acceptance. Treatment with Rituxan has brought relief to many patients whose synovitis symptoms, including pain and stiffness, subside, as do the swollen hand joints that accompany the more advanced stages of the disease. Today treating rheumatoid arthritis with anti-CD20 blockers is well accepted, but this was not the case a few short years ago.
The transformation in thinking began in 1997 when Rituxan was approved for blood cancers. As the drug became accessible to investigators like Cambridge and Edwards, they were able to test the idea that B cells were driving autoimmune disease. This was anathema at the time because most research in autoimmunity focused on T cells. The confusion is understandable because B cells and T cells signal back and forth continuously in a highly integrated and dynamic network. B cells present antigens to T cells, which in turn activate and excite B cells to produce autoantibodies such as rheumatoid factor. These RF antibodies latch on to the Fc portions of IgGs to form immune complexes, which in turn trigger a cascade of events that finally attract macrophages and neutrophils. As these phagocytic cells engulf immunocomplexes, they release lysosomal enzymes that ratchet up inflammation and further damage the synovial apparatus, leading to chronic disease.
Anti-CD20 agent Rituxan interrupts this self-perpetuating cycle by wiping out B-cell populations. This may seem an extreme measure, but it has allowed a sizable proportion of rheumatoid arthritis patients to manage their disease. Not all CD20 blockers are equivalent, however. CD20 is a tetra-membrane-spanning protein with a rather serpentine configuration weaving in and out of the B-cell membrane. Different anti-CD20 antibodies might bind epitopes far apart on the molecule, and these differences could account for variations in intracellular signaling cascades affecting potency, efficacy or even immunotoxicity. Different antibody formats may also have different advantages in terms of functionality, pharmacodynamics and pharmacokinetics.
One novel antibody format targeting CD20 currently under development is Seattle-based Trubion Pharmaceuticals' TRU-015. This molecule—one third of the size of a true antibody—consists of single-chain variable regions (VL and VH) that bind CD20, which are fused by means of a modified human IgG1 hinge domain to engineered constant regions that encode human IgG1 constant heavy domains (CH2 and CH3). Because the fusion protein includes the Fc portion it allows antibody-dependent cellular cytotoxicity (ADCC) to take place whereas its smaller size is thought to improve tissue biodistribution.
In collaboration with Madison, New Jersey-based Wyeth, Trubion is studying this anti-CD20 scFv-Fc fusion protein (TRU-015) in phase 2 trials for rheumatoid arthritis and in preclinical development for systemic lupus erythematosus (SLE). CEO and founder Peter Thompson says the product is "significantly attenuated" with respect to complement-dependent cellular cytotoxicity (CDCC) of B cells, but "very potent" with respect to ADCC and direct apoptotic activity (these are two of the mechanisms by which existing anti-CD20 antibodies are thought to deplete B cells). Such toned-down CDCC activity, relative to Rituxan, is presumably advantageous because complement cascades have been shown to herald signs and symptoms of some infusion reactions, which can range from very mild to severe. "We have not really seen grade three or grade four infusion reactions with TRU-015 in contrast to the experience previously published with Rituxan," says Thompson. Trubion also has another scFv-Fc fusion (SBI-087) targeting CD20 in phase 1 trials for SLE and rheumatoid arthritis.
In the early days of Rituxan as a rheumatoid arthritis treatment, rheumatologists were concerned over what might happen if the patient's B-cell population were to be wiped out, and if their immunoglobulin levels were to drop to unacceptably low levels. Some of these worries have now been dispelled. B cells give rise to long-lived immunoglobulin-producing plasma cells that persist for many months, and because they do not have the CD20 antigen on their cell surfaces, these plasma cells remain untouched by anti-CD20 agents. Also, plasma cells retain some memory of previous exposures to infectious agents, so they continue to offer protection to the patient. "Patients treated with rituximab [Rituxan] still mount an immunoglobulin response to vaccines after rituximab therapy," says rheumatologist Peter Taylor, who is head of clinical trials at the Kennedy Institute of Rheumatology in London. "But humoral responses can be reduced," he says.
The critical question for clinicians is what happens to the patients' bone marrow stem cells and their ability to generate plasma cells after repeated cycles of anti-CD20 therapy. As bone marrow precursors are not CD20 positive, presumably they are not affected by anti-CD20 agents. "In my own practice, we've gotten up to four or five cycles," says Taylor. Cambridge and Edwards have doubled the cycles without observing substantial drops in immunoglobulin levels. They believe that after B cells in the periphery are depleted, the bone marrow somehow reboots to produce new lymphocytes that no longer churn out rheumatoid arthritis–causing autoantibodies. Indeed, Cambridge has been tracking a group of rheumatoid arthritis patients whose symptoms have not returned for 18 months after the last Rituxan treatment. Cambridge won't go out on a limb and say there's been a complete response, but she says, "One patient has been in remission for three years or longer."
Still, Taylor expresses some mild concerns. "When you get past about five or six cycles of therapy, there is a proportion of patients with robust and well-maintained, really good responses, that at some point don't recover B-cell levels, and their immunoglobulin levels drop down below the normal range," he says. "If B-cell depletion were thought to be the optimal therapy for individuals with rheumatoid arthritis, it may have a finite shelf life as to how long you could use that approach. It may be perhaps that you could use rituximab [Rituxan] in a responsive patient for 5 years, 10 years or 12 years. But it's not yet clear that you could go on [long term], as we do, with methotrexate for a lifetime."
Time will tell if newer anti-CD20 therapies in pipelines work any better. Because ofatumumab is fully human and Rituxan is chimeric, the supposition is that ofatumumab would present fewer infusion reactions or other adverse events, particularly the human anti-chimeric antibody response. But not much information is publicly available, and, in fact, there have been no head-to-head trials of ofatumumab versus Rituxan in rheumatoid arthritis. Taylor is principal investigator in one ongoing phase 3 trial of ofatumumab for rheumatoid arthritis, which is scheduled to wrap up in 2011, but a confidentiality agreement prevents him from discussing it.
One thing Taylor will say, however, is that there's been a revolution in the mind-set of rheumatologists around the globe, who are losing their trepidation about using cancer drugs in rheumatoid arthritis patients. "I think there's a massive change," he says. "People often use the phrase 'aggressive early therapy'. To me, it really means optimally suppressing synovitis."
mfg ipollit
http://www.nature.com/nbt/journal/v26/n10/full/nbt1008-1053.…
Nature Biotechnology 26, 1053 - 1054 (2008)
doi:10.1038/nbt1008-1053
CD20 blockers eye crowded rheumatology market
George S Mack1
Columbia, South Carolina
When trial results for a novel cancer drug were trumpeted in July, the rheumatology field felt the ripples. The drug is ofatumumab, a monoclonal antibody (mAb) targeting the CD20 molecule on B lymphocytes. Its makers, London-based GlaxoSmithKline (GSK) and Genmab of Copenhagen, announced that the mAb had met its primary and secondary endpoints in chronic lymphocytic leukemia (CLL). But with two double-blind phase 3 studies of ofatumumab for rheumatoid arthritis already underway in Italy and Eastern Europe, these companies are clearly looking beyond cancer indications to lucrative autoimmune disease markets for their second-generation molecule.
The idea is to follow the trail blazed by Genentech of South San Francisco, Roche of Basel and Biogen-Idec of Cambridge, Massachusetts, with Rituxan (rituximab), their anti-CD20 chimeric mAb. Rituxan was approved by the US Food and Drug Administration to treat B-cell lymphomas and leukemias in 1997 and then as a third-line therapy against rheumatoid arthritis in 2006. Currently marketed by Roche in Europe as MabThera, it surpassed $5 billion in worldwide sales in 2007 alone.
GSK has invested heavily in ofatumumab. The company made headlines at the end of 2006 when the licensing agreement to co-develop this fully human IgG1- mAb for cancer and rheumatoid arthritis was announced. The deal, potentially worth $2.1 billion, saw Genmab receive an upfront $102 million as a sweetener for their anti-CD20 mAb. GSK also took a 10% stake in the biotech firm by purchasing $357 million in shares. The transaction was deemed to be the largest ever in Europe for a single product; if all milestones are met it will exceed the $2.0 billion deal between New York–based Bristol-Myers Squibb and New York–based ImClone Systems for blockbuster cancer chimeric mAb Erbitux (cetuximab).
The meteoric rise of tumor necrosis factor (TNF)- blockers over the past decade ushered in the age of biologic therapies for rheumatoid arthritis. Anti-TNF agents—Enbrel (etanercept), Humira (adalimumab) and Remicade (infliximab)—are now labeled for second-line use after failure of methotrexate and nonsteroidal anti-inflammatory drugs (NSAIDs). But there is no product that works for everyone—about a third of those with rheumatoid arthritis do not respond to first- or second-line therapy.
Rituxan is currently a third-line therapy for rheumatoid arthritis after failure of methotrexate and anti-TNF- products. "We'd like to try it in patients much earlier on, and we're working towards that at the moment," says immunologist Geraldine Cambridge, who has collaborated for many years with rheumatologist Jonathan Edwards, both of the Center for Rheumatology Research at University College London. The two have done much of the basic science groundwork as well as overseen clinical trials over the past decade to establish the effectiveness of B-cell depletion with CD20 blockade. "You need to get these patients in remission as early as possible," she says. "If you lose one year while patients are slowly failing on methotrexate and TNF-inhibitors, that means they've lost their job by then and will probably never get back to work again."
The clinical approach by which B cells are ablated to treat rheumatoid arthritis has only recently gained acceptance. Treatment with Rituxan has brought relief to many patients whose synovitis symptoms, including pain and stiffness, subside, as do the swollen hand joints that accompany the more advanced stages of the disease. Today treating rheumatoid arthritis with anti-CD20 blockers is well accepted, but this was not the case a few short years ago.
The transformation in thinking began in 1997 when Rituxan was approved for blood cancers. As the drug became accessible to investigators like Cambridge and Edwards, they were able to test the idea that B cells were driving autoimmune disease. This was anathema at the time because most research in autoimmunity focused on T cells. The confusion is understandable because B cells and T cells signal back and forth continuously in a highly integrated and dynamic network. B cells present antigens to T cells, which in turn activate and excite B cells to produce autoantibodies such as rheumatoid factor. These RF antibodies latch on to the Fc portions of IgGs to form immune complexes, which in turn trigger a cascade of events that finally attract macrophages and neutrophils. As these phagocytic cells engulf immunocomplexes, they release lysosomal enzymes that ratchet up inflammation and further damage the synovial apparatus, leading to chronic disease.
Anti-CD20 agent Rituxan interrupts this self-perpetuating cycle by wiping out B-cell populations. This may seem an extreme measure, but it has allowed a sizable proportion of rheumatoid arthritis patients to manage their disease. Not all CD20 blockers are equivalent, however. CD20 is a tetra-membrane-spanning protein with a rather serpentine configuration weaving in and out of the B-cell membrane. Different anti-CD20 antibodies might bind epitopes far apart on the molecule, and these differences could account for variations in intracellular signaling cascades affecting potency, efficacy or even immunotoxicity. Different antibody formats may also have different advantages in terms of functionality, pharmacodynamics and pharmacokinetics.
One novel antibody format targeting CD20 currently under development is Seattle-based Trubion Pharmaceuticals' TRU-015. This molecule—one third of the size of a true antibody—consists of single-chain variable regions (VL and VH) that bind CD20, which are fused by means of a modified human IgG1 hinge domain to engineered constant regions that encode human IgG1 constant heavy domains (CH2 and CH3). Because the fusion protein includes the Fc portion it allows antibody-dependent cellular cytotoxicity (ADCC) to take place whereas its smaller size is thought to improve tissue biodistribution.
In collaboration with Madison, New Jersey-based Wyeth, Trubion is studying this anti-CD20 scFv-Fc fusion protein (TRU-015) in phase 2 trials for rheumatoid arthritis and in preclinical development for systemic lupus erythematosus (SLE). CEO and founder Peter Thompson says the product is "significantly attenuated" with respect to complement-dependent cellular cytotoxicity (CDCC) of B cells, but "very potent" with respect to ADCC and direct apoptotic activity (these are two of the mechanisms by which existing anti-CD20 antibodies are thought to deplete B cells). Such toned-down CDCC activity, relative to Rituxan, is presumably advantageous because complement cascades have been shown to herald signs and symptoms of some infusion reactions, which can range from very mild to severe. "We have not really seen grade three or grade four infusion reactions with TRU-015 in contrast to the experience previously published with Rituxan," says Thompson. Trubion also has another scFv-Fc fusion (SBI-087) targeting CD20 in phase 1 trials for SLE and rheumatoid arthritis.
In the early days of Rituxan as a rheumatoid arthritis treatment, rheumatologists were concerned over what might happen if the patient's B-cell population were to be wiped out, and if their immunoglobulin levels were to drop to unacceptably low levels. Some of these worries have now been dispelled. B cells give rise to long-lived immunoglobulin-producing plasma cells that persist for many months, and because they do not have the CD20 antigen on their cell surfaces, these plasma cells remain untouched by anti-CD20 agents. Also, plasma cells retain some memory of previous exposures to infectious agents, so they continue to offer protection to the patient. "Patients treated with rituximab [Rituxan] still mount an immunoglobulin response to vaccines after rituximab therapy," says rheumatologist Peter Taylor, who is head of clinical trials at the Kennedy Institute of Rheumatology in London. "But humoral responses can be reduced," he says.
The critical question for clinicians is what happens to the patients' bone marrow stem cells and their ability to generate plasma cells after repeated cycles of anti-CD20 therapy. As bone marrow precursors are not CD20 positive, presumably they are not affected by anti-CD20 agents. "In my own practice, we've gotten up to four or five cycles," says Taylor. Cambridge and Edwards have doubled the cycles without observing substantial drops in immunoglobulin levels. They believe that after B cells in the periphery are depleted, the bone marrow somehow reboots to produce new lymphocytes that no longer churn out rheumatoid arthritis–causing autoantibodies. Indeed, Cambridge has been tracking a group of rheumatoid arthritis patients whose symptoms have not returned for 18 months after the last Rituxan treatment. Cambridge won't go out on a limb and say there's been a complete response, but she says, "One patient has been in remission for three years or longer."
Still, Taylor expresses some mild concerns. "When you get past about five or six cycles of therapy, there is a proportion of patients with robust and well-maintained, really good responses, that at some point don't recover B-cell levels, and their immunoglobulin levels drop down below the normal range," he says. "If B-cell depletion were thought to be the optimal therapy for individuals with rheumatoid arthritis, it may have a finite shelf life as to how long you could use that approach. It may be perhaps that you could use rituximab [Rituxan] in a responsive patient for 5 years, 10 years or 12 years. But it's not yet clear that you could go on [long term], as we do, with methotrexate for a lifetime."
Time will tell if newer anti-CD20 therapies in pipelines work any better. Because ofatumumab is fully human and Rituxan is chimeric, the supposition is that ofatumumab would present fewer infusion reactions or other adverse events, particularly the human anti-chimeric antibody response. But not much information is publicly available, and, in fact, there have been no head-to-head trials of ofatumumab versus Rituxan in rheumatoid arthritis. Taylor is principal investigator in one ongoing phase 3 trial of ofatumumab for rheumatoid arthritis, which is scheduled to wrap up in 2011, but a confidentiality agreement prevents him from discussing it.
One thing Taylor will say, however, is that there's been a revolution in the mind-set of rheumatologists around the globe, who are losing their trepidation about using cancer drugs in rheumatoid arthritis patients. "I think there's a massive change," he says. "People often use the phrase 'aggressive early therapy'. To me, it really means optimally suppressing synovitis."
mfg ipollit
Cubist... Cubicin entwickelt sich weiter sehr positiv
Cubist Pharma Q3 profit surges 39% on Cubicin sales - Update
10/16/2008 11:17 PM ET
(RTTNews) - Thursday, biopharmaceutical company Cubist Pharmaceuticals, Inc. (CBST: News ) reported a 39% surge in profit for the third quarter on strong sales of its antibiotic Cubicin. Total revenue for the quarter rose 41%.
The Lexington, Massachusetts-based company's net income on a GAAP basis increased to $27.93 million, or $0.44 per share, from $20.02 million, or $0.32 per share, in the same quarter last year.
The company said results for the latest quarter included $2.9 million, or $0.04 per share, net of tax, in stock-based compensation expenses.
Excluding stock-based compensation expense, the company's non-GAAP net income for the quarter increased to $30.82 million, or $0.48 per share, from $22.50 million, or $0.36 per share, in the same quarter of the previous year.
On average, thirteen analysts polled by First Call/Thomson Financials estimated the company to earn $0.33 per share for the quarter.
Total revenues for the quarter rose 41% to $112.44 million from $79.80 million in the prior year quarter. Wall Street analysts expected the company to post revenues for the quarter of $108.74 million.
Revenue from Cubicin surged 45% to $110.6 million from $76.3 million in the same period last year. U.S. net product revenue from Cubicin rose to $109.20 million from $75.37 million in the year-ago period.
In addition, revenue for the latest quarter includes $1.4 million of service revenue relating to Cubist's exclusive agreement with AstraZeneca to sell and provide other support in the U.S. for antibiotic Merrem I.V.
Cubist's operating income for the quarter increased to $28.72 million from $18.25 million in the same period last year.
For the nine-month period, the company's net income rose to $46.78 million, or $0.75 per share from $40.11 million, or $0.68 per share in the same period of the prior year.
Non-GAAP pro forma net income for the period rose to $74.69 million, or $1.16 per share, from $47.75 million, or $0.79 per share, in the same period of the previous year.
Total revenues for the nine months were $302.49 million, up from $209.04 million in the corresponding period of the previous year.
mfg ipollit
Cubist Pharma Q3 profit surges 39% on Cubicin sales - Update
10/16/2008 11:17 PM ET
(RTTNews) - Thursday, biopharmaceutical company Cubist Pharmaceuticals, Inc. (CBST: News ) reported a 39% surge in profit for the third quarter on strong sales of its antibiotic Cubicin. Total revenue for the quarter rose 41%.
The Lexington, Massachusetts-based company's net income on a GAAP basis increased to $27.93 million, or $0.44 per share, from $20.02 million, or $0.32 per share, in the same quarter last year.
The company said results for the latest quarter included $2.9 million, or $0.04 per share, net of tax, in stock-based compensation expenses.
Excluding stock-based compensation expense, the company's non-GAAP net income for the quarter increased to $30.82 million, or $0.48 per share, from $22.50 million, or $0.36 per share, in the same quarter of the previous year.
On average, thirteen analysts polled by First Call/Thomson Financials estimated the company to earn $0.33 per share for the quarter.
Total revenues for the quarter rose 41% to $112.44 million from $79.80 million in the prior year quarter. Wall Street analysts expected the company to post revenues for the quarter of $108.74 million.
Revenue from Cubicin surged 45% to $110.6 million from $76.3 million in the same period last year. U.S. net product revenue from Cubicin rose to $109.20 million from $75.37 million in the year-ago period.
In addition, revenue for the latest quarter includes $1.4 million of service revenue relating to Cubist's exclusive agreement with AstraZeneca to sell and provide other support in the U.S. for antibiotic Merrem I.V.
Cubist's operating income for the quarter increased to $28.72 million from $18.25 million in the same period last year.
For the nine-month period, the company's net income rose to $46.78 million, or $0.75 per share from $40.11 million, or $0.68 per share in the same period of the prior year.
Non-GAAP pro forma net income for the period rose to $74.69 million, or $1.16 per share, from $47.75 million, or $0.79 per share, in the same period of the previous year.
Total revenues for the nine months were $302.49 million, up from $209.04 million in the corresponding period of the previous year.
mfg ipollit
Antwort auf Beitrag Nr.: 35.613.851 von ipollit am 18.10.08 15:58:01CBST...
http://seekingalpha.com/article/100349-cubist-pharmaceutical…
Cubist Pharmaceuticals Inc. Q3 2008 Earnings Call Transcript
...
Our top line is fueled by the continued growth of CUBICIN, which, in Q3, achieved year-to-year net revenue growth in the US of 45%. This level of year-to-year growth is particularly impressive as CUBICIN has now passed the five-year anniversary of its initial approval in the US.
This quarter, we are reporting for the first time a contribution from MERREM, which we began selling for AstraZeneca in US hospitals in late July. We are very pleased by the results here. As you will recall, based on GAAP accounting rules, the lion share of the service revenues we expect to earn for selling MERREM in 2008 will be reflected in Q4 results.
Steve will be providing some important updates on our advancing Phase 2 candidate ecallantide, being developed for the reduction of blood loss during on-pump cardiothoracic surgery, or CTS.
The unmet medical need here is quite significant and no one else has a product as far along in development as ecallantide for blood loss in cardiothoracic surgery. We estimate a market opportunity for this drug of greater than $500 million. We also are on target for two IND filings by the end of this year for our Cubist discovered antibiotic candidates, one for the treatment of multi-drug resistant Gram-negative infections and the second for the treatment of C-difficile infections.
We continue to make slow but steady progress against our main competitor, vancomycin. For the year-to-date period through August versus the same period last year, vancomycin has lost 1.5 share points while CUBICIN has increased its share by 1.9 points. As a result, we have narrowed the gap between CUBICIN and vancomycin by almost 3.5 share points thus far in 2008.
In our Phase 2 ecallantide program for reducing blood loss in patients undergoing on-pump cardiothoracic surgery, we have had a very positive and productive quarter of activity.
Our initial interest in licensing ecallantide was due, in part, to the results of a relatively small proof-of-concept trial in 41 patients, known as the 883 trial in which ecallantide treatment of patients undergoing primary CABG surgery resulted in approximately 50% reduction in transfusion volumes, compared to placebo. In addition, the Kalahari 1 trial was underway which was designed to evaluate the safety and efficacy of a low and a high dose of ecallantide.
The exciting news from Kalahari 1 with a total of 69 patients dosed is that the efficacy signals seen was consistent with that observed in the smaller 883 proof-of-concept trial.
Also this quarter, we have had very positive interactions with our advisory board of experts in the cardiac surgery field regarding the protocol design for the Phase 2 dose ranging trial.
In this trial, we will study the safety and efficacy of two or three doses of ecallantide compared with placebo in primary CABG patients, using transfusion volume as the key efficacy parameter. In addition, we now also plan to proceed with a second parallel Phase 2 trial in a surgical population at higher risk of bleeding which will help us generate information on the safety and efficacy of ecallantide treatment in this population prior to Phase 3 studies.
Protocols for both trials are now under review internally and with our key opinion leader advisory board. We expect to launch the dose ranging trial by year end, followed shortly by the start of the trial in higher risk patients. We anticipate that these two trials, which we expect to complete in early 2010, will together provide a comprehensive data set on approximately 500 patients for our end-of-Phase 2 meeting with the FDA, which we are targeting for mid-2010.
Total revenues of $112.4 million are up 41% from Q3 2007, and include for the first time MERREM service revenue of $1.4 million. US net CUBICIN revenue of $109.2 for Q3 2008 is up 45% from Q3 2007. Gross margin for Q3 was 78.7%, which is up from 77.5% in Q3 2007.
Net income of $27.9 million is up 40% over Q3 2007 and 30% of the increase in gross margin year-over-year has dropped to the bottom line. EPS fully diluted is $0.44.
Our cash equivalents and investments were $373 million at quarter end, which is up $15 million from the quarter end Q2.
Now to guidance. We are increasing the US net revenue guidance to a range of $410 million to $420 million.
...
mfg ipollit
http://seekingalpha.com/article/100349-cubist-pharmaceutical…
Cubist Pharmaceuticals Inc. Q3 2008 Earnings Call Transcript
...
Our top line is fueled by the continued growth of CUBICIN, which, in Q3, achieved year-to-year net revenue growth in the US of 45%. This level of year-to-year growth is particularly impressive as CUBICIN has now passed the five-year anniversary of its initial approval in the US.
This quarter, we are reporting for the first time a contribution from MERREM, which we began selling for AstraZeneca in US hospitals in late July. We are very pleased by the results here. As you will recall, based on GAAP accounting rules, the lion share of the service revenues we expect to earn for selling MERREM in 2008 will be reflected in Q4 results.
Steve will be providing some important updates on our advancing Phase 2 candidate ecallantide, being developed for the reduction of blood loss during on-pump cardiothoracic surgery, or CTS.
The unmet medical need here is quite significant and no one else has a product as far along in development as ecallantide for blood loss in cardiothoracic surgery. We estimate a market opportunity for this drug of greater than $500 million. We also are on target for two IND filings by the end of this year for our Cubist discovered antibiotic candidates, one for the treatment of multi-drug resistant Gram-negative infections and the second for the treatment of C-difficile infections.
We continue to make slow but steady progress against our main competitor, vancomycin. For the year-to-date period through August versus the same period last year, vancomycin has lost 1.5 share points while CUBICIN has increased its share by 1.9 points. As a result, we have narrowed the gap between CUBICIN and vancomycin by almost 3.5 share points thus far in 2008.
In our Phase 2 ecallantide program for reducing blood loss in patients undergoing on-pump cardiothoracic surgery, we have had a very positive and productive quarter of activity.
Our initial interest in licensing ecallantide was due, in part, to the results of a relatively small proof-of-concept trial in 41 patients, known as the 883 trial in which ecallantide treatment of patients undergoing primary CABG surgery resulted in approximately 50% reduction in transfusion volumes, compared to placebo. In addition, the Kalahari 1 trial was underway which was designed to evaluate the safety and efficacy of a low and a high dose of ecallantide.
The exciting news from Kalahari 1 with a total of 69 patients dosed is that the efficacy signals seen was consistent with that observed in the smaller 883 proof-of-concept trial.
Also this quarter, we have had very positive interactions with our advisory board of experts in the cardiac surgery field regarding the protocol design for the Phase 2 dose ranging trial.
In this trial, we will study the safety and efficacy of two or three doses of ecallantide compared with placebo in primary CABG patients, using transfusion volume as the key efficacy parameter. In addition, we now also plan to proceed with a second parallel Phase 2 trial in a surgical population at higher risk of bleeding which will help us generate information on the safety and efficacy of ecallantide treatment in this population prior to Phase 3 studies.
Protocols for both trials are now under review internally and with our key opinion leader advisory board. We expect to launch the dose ranging trial by year end, followed shortly by the start of the trial in higher risk patients. We anticipate that these two trials, which we expect to complete in early 2010, will together provide a comprehensive data set on approximately 500 patients for our end-of-Phase 2 meeting with the FDA, which we are targeting for mid-2010.
Total revenues of $112.4 million are up 41% from Q3 2007, and include for the first time MERREM service revenue of $1.4 million. US net CUBICIN revenue of $109.2 for Q3 2008 is up 45% from Q3 2007. Gross margin for Q3 was 78.7%, which is up from 77.5% in Q3 2007.
Net income of $27.9 million is up 40% over Q3 2007 and 30% of the increase in gross margin year-over-year has dropped to the bottom line. EPS fully diluted is $0.44.
Our cash equivalents and investments were $373 million at quarter end, which is up $15 million from the quarter end Q2.
Now to guidance. We are increasing the US net revenue guidance to a range of $410 million to $420 million.
...
mfg ipollit
Antwort auf Beitrag Nr.: 35.613.969 von ipollit am 18.10.08 16:13:37Cubist: eps-Schätzungen liegen aktuell laut yahoo bei 1,24 USD und 1,73 USD für 2008 bzw. 2009. Dies entspricht bei dem aktuellen Kurs von 21,64 USD einem KGVe von 17,4 bzw. 12,5. Der Cash liegt laut CC bei 373 Mio USD bei einer aktuellen MK von 1,22 Mrd USD und positivem Cashflow.
mfg ipollit
mfg ipollit
Antwort auf Beitrag Nr.: 35.613.358 von ipollit am 18.10.08 14:55:42ein paar Folien zu Genmab's HuMax-CD20 vs. Rituxan







mfg ipollit
mfg ipollit
Exelixis... BMS kann jetzt mit XL413 neben XL139 einen weiteren Kandidaten von EXEL in seine Pipeline übernehmen. Die Entscheidung darüber fällt in der nächsten Zeit.
Exelixis Submits Diligence Report for Cdc7 Inhibitor to Bristol-Myers Squibb
Second IND Candidate Advances for Potential Co-Development Opportunity in Oncology Collaboration
Last update: 4:05 p.m. EDT Oct. 15, 2008
SOUTH SAN FRANCISCO, Calif., Oct 15, 2008 (BUSINESS WIRE) -- Exelixis, Inc. (EXEL) today announced that it has submitted a comprehensive data report for investigational new drug (IND) candidate XL413, a selective inhibitor of Cdc7, to Bristol-Myers Squibb Company (BMY) . Bristol-Myers Squibb now has 30 days to review the data package and determine if it will select the compound for clinical development and commercialization. If Bristol-Myers Squibb selects XL413, Exelixis will be entitled to a $20 million milestone payment under the 2007 collaboration agreement with Bristol-Myers Squibb to discover, develop, and commercialize novel targeted therapies for the treatment of cancer.
"XL413 is the second compound that we have submitted under our current oncology discovery collaboration with Bristol-Myers Squibb," said Michael Morrissey, PhD, President of Research and Development at Exelixis. "One of our key R&D goals is to continue to expand our pipeline with new compounds focused on key pathways and targets that play important roles in cancer. XL413 selectively targets Cdc7, which is a very exciting cancer-related target. To our knowledge, no other selective inhibitors of Cdc7 have advanced to this stage of preclinical development, giving XL413 potential to become a first-in-class therapy."
About XL413
XL413 is a small molecule inhibitor of the serine-threonine kinase Cdc7. The function of Cdc7 is required for DNA replication to proceed, and its activity is often upregulated in cancer cells. Studies suggest that Cdc7 plays a role in regulation of cell cycle checkpoint control and protects tumor cells from apoptotic cell death during replication stress. Therefore, inhibition of Cdc7 may have utility in the treatment of a wide variety of cancers, either as a single agent or in combination with DNA damaging agents.
Background on the Collaboration Agreement
Under the terms of the agreement, Exelixis is fully responsible for the identification and preclinical development of small molecule drug candidates directed against mutually selected targets. In addition to an upfront payment of $60 million in January 2007, the agreement provides for Exelixis to receive $20 million for each of up to three different IND-stage drug candidates selected by Bristol-Myers Squibb. For each candidate selected by Bristol-Myers Squibb, Exelixis has the right to opt in to the co-development or co-promotion in the United States. If Exelixis does not opt in to co-promote the selected IND candidates, Exelixis would be entitled to receive milestones and royalties in lieu of profits from sales in the United States. Outside of the United States, Bristol-Myers Squibb will have primary responsibility for development activities and Exelixis will be entitled to receive royalties on product sales.
In January 2008, Bristol-Myers Squibb selected XL139, a small molecule inhibitor of the hedgehog signaling pathway, for further development and commercialization. In connection with this selection, Exelixis received a $20 million milestone payment and also exercised its option under the collaboration agreement to co-develop and co-commercialize XL139 in the United States. Following the transfer of the XL139 development program in mid-2008, Bristol-Myers Squibb is leading all global activities.
mfg ipollit
Exelixis Submits Diligence Report for Cdc7 Inhibitor to Bristol-Myers Squibb
Second IND Candidate Advances for Potential Co-Development Opportunity in Oncology Collaboration
Last update: 4:05 p.m. EDT Oct. 15, 2008
SOUTH SAN FRANCISCO, Calif., Oct 15, 2008 (BUSINESS WIRE) -- Exelixis, Inc. (EXEL) today announced that it has submitted a comprehensive data report for investigational new drug (IND) candidate XL413, a selective inhibitor of Cdc7, to Bristol-Myers Squibb Company (BMY) . Bristol-Myers Squibb now has 30 days to review the data package and determine if it will select the compound for clinical development and commercialization. If Bristol-Myers Squibb selects XL413, Exelixis will be entitled to a $20 million milestone payment under the 2007 collaboration agreement with Bristol-Myers Squibb to discover, develop, and commercialize novel targeted therapies for the treatment of cancer.
"XL413 is the second compound that we have submitted under our current oncology discovery collaboration with Bristol-Myers Squibb," said Michael Morrissey, PhD, President of Research and Development at Exelixis. "One of our key R&D goals is to continue to expand our pipeline with new compounds focused on key pathways and targets that play important roles in cancer. XL413 selectively targets Cdc7, which is a very exciting cancer-related target. To our knowledge, no other selective inhibitors of Cdc7 have advanced to this stage of preclinical development, giving XL413 potential to become a first-in-class therapy."
About XL413
XL413 is a small molecule inhibitor of the serine-threonine kinase Cdc7. The function of Cdc7 is required for DNA replication to proceed, and its activity is often upregulated in cancer cells. Studies suggest that Cdc7 plays a role in regulation of cell cycle checkpoint control and protects tumor cells from apoptotic cell death during replication stress. Therefore, inhibition of Cdc7 may have utility in the treatment of a wide variety of cancers, either as a single agent or in combination with DNA damaging agents.
Background on the Collaboration Agreement
Under the terms of the agreement, Exelixis is fully responsible for the identification and preclinical development of small molecule drug candidates directed against mutually selected targets. In addition to an upfront payment of $60 million in January 2007, the agreement provides for Exelixis to receive $20 million for each of up to three different IND-stage drug candidates selected by Bristol-Myers Squibb. For each candidate selected by Bristol-Myers Squibb, Exelixis has the right to opt in to the co-development or co-promotion in the United States. If Exelixis does not opt in to co-promote the selected IND candidates, Exelixis would be entitled to receive milestones and royalties in lieu of profits from sales in the United States. Outside of the United States, Bristol-Myers Squibb will have primary responsibility for development activities and Exelixis will be entitled to receive royalties on product sales.
In January 2008, Bristol-Myers Squibb selected XL139, a small molecule inhibitor of the hedgehog signaling pathway, for further development and commercialization. In connection with this selection, Exelixis received a $20 million milestone payment and also exercised its option under the collaboration agreement to co-develop and co-commercialize XL139 in the United States. Following the transfer of the XL139 development program in mid-2008, Bristol-Myers Squibb is leading all global activities.
mfg ipollit
Exelixis, Array und Seattle Genetics...
http://www.fool.com/investing/high-growth/2008/10/01/3-drugm…
3 Drugmakers With Multiple Shots on Goal
By Brian Orelli
October 1, 2008
Investing in development-stage drugmakers is risky. Really risky. One setback and the company's stock can plummet. Heck, even without setbacks, the stock can head downward. There are two ways to mitigate that risk. The first is to buy a lot of drug companies or keep them as a small fraction of your portfolio.
The second way is to buy companies with multiple chances of getting a drug approved. That way, if one drug fails in clinical trials, the company isn't as likely to tank as much as a company with just one major prospect would. Using either of these strategies could help you hit a home run with your biotech investments.
Today, I'm going to take a look at three companies that address that second factor. Each of these companies has pretty deep pipelines, but no approved drugs yet.
Company
Drugs in Phase 1
Drugs in Phase 2
Drugs in Phase 3
Total
Exelixis (Nasdaq: EXEL)
7
3
1
11
Seattle Genetics (Nasdaq: SGEN)
2
2
0
4
Array BioPharma (Nasdaq: ARRY)
3
4
0
7
Now that we see where these three stand, what are their prospects?
So many candidates, so little cash
Exelixis is a drug discovery machine. It has an uncanny ability to discover inhibitors of kinases, key control points for many different processes in the cell. The company should be able to develop a diverse offering.
With a goal of adding three new compounds to the pipeline each year, Exelixis has no worries about having its pipeline run dry. On the contrary, it has so many drug candidates that it can't possibly run them all through phase 3 development with its cash on hand, only $159 million.
Fortunately it has partnerships with Genentech (NYSE: DNA) and GlaxoSmithKline (NYSE: GSK). Glaxo has already picked up one of Exelixis' 11 drugs and has an option to pick up another.
But one of its phase 2 drugs, non-small cell lung cancer treatment XL647, is being shelved because Exelixis doesn't have the cash to invest in the drug. Lung cancer is a highly competitive market, and paying for a phase 3 trial wouldn't necessarily be the best use of the company's limited resources.
Antibody platform
Seattle Genetics has based its research on antibodies -- specifically antibodies that can kill cancer cells. Some of the antibodies are engineered to directly kill the cells, similar to the way Genentech's Avastin or ImClone Systems' (Nasdaq: IMCL) Erbitux work. But it is Seattle Genetics' indirect killing mechanism that probably has the best chance of stocking its pipeline.
The company has developed a way to add a toxic drug to an antibody to send the drug specifically to the cancer cells. The beauty of the system is that it can be added to a large number of different antibodies, depending on what types of cells the drug is trying to kill.
Not surprisingly, larger drug companies have become very interested in the system and have licensed Seattle Genetics' technology to develop their own antibody drugs. You can almost hear the cha-ching of royalty payments coming.
But that's not all
The seven compounds in Array's pipeline may look small and immature, but the company doesn't count its partnerships on its main pipeline page. Its phase 2 drug ARRY-886 is partnered with AstraZeneca (NYSE: AZN), and although it showed underwhelming results by itself, the drug may work better in combination with other chemotherapies. Array also has partnered with InterMune on hepatitis C drug ITMN-191, which had pretty good-looking phase 1 clinical trial results.
No such thing as a sure thing
These drug developers are still far from a sure thing. They could run out of money -- or dilute shareholder value by doing many secondary offerings -- and turn out to be lousy investments. But the fact that they all have well-stocked pipelines gives them an edge over other drugmakers.
And if they're able to get just one drug approved, then watch out, because their stock is likely to take off like a rocket. All you have to do is look at the market cap of a one-drug wonder like Onyx Pharmaceuticals or Amylin Pharmaceuticals to realize that these developmental-stage drugmakers have a long way to run if they can get just a single successful drug approved. And that's what makes investing in biotech so much fun.
mfg ipollit
http://www.fool.com/investing/high-growth/2008/10/01/3-drugm…
3 Drugmakers With Multiple Shots on Goal
By Brian Orelli
October 1, 2008
Investing in development-stage drugmakers is risky. Really risky. One setback and the company's stock can plummet. Heck, even without setbacks, the stock can head downward. There are two ways to mitigate that risk. The first is to buy a lot of drug companies or keep them as a small fraction of your portfolio.
The second way is to buy companies with multiple chances of getting a drug approved. That way, if one drug fails in clinical trials, the company isn't as likely to tank as much as a company with just one major prospect would. Using either of these strategies could help you hit a home run with your biotech investments.
Today, I'm going to take a look at three companies that address that second factor. Each of these companies has pretty deep pipelines, but no approved drugs yet.
Company
Drugs in Phase 1
Drugs in Phase 2
Drugs in Phase 3
Total
Exelixis (Nasdaq: EXEL)
7
3
1
11
Seattle Genetics (Nasdaq: SGEN)
2
2
0
4
Array BioPharma (Nasdaq: ARRY)
3
4
0
7
Now that we see where these three stand, what are their prospects?
So many candidates, so little cash
Exelixis is a drug discovery machine. It has an uncanny ability to discover inhibitors of kinases, key control points for many different processes in the cell. The company should be able to develop a diverse offering.
With a goal of adding three new compounds to the pipeline each year, Exelixis has no worries about having its pipeline run dry. On the contrary, it has so many drug candidates that it can't possibly run them all through phase 3 development with its cash on hand, only $159 million.
Fortunately it has partnerships with Genentech (NYSE: DNA) and GlaxoSmithKline (NYSE: GSK). Glaxo has already picked up one of Exelixis' 11 drugs and has an option to pick up another.
But one of its phase 2 drugs, non-small cell lung cancer treatment XL647, is being shelved because Exelixis doesn't have the cash to invest in the drug. Lung cancer is a highly competitive market, and paying for a phase 3 trial wouldn't necessarily be the best use of the company's limited resources.
Antibody platform
Seattle Genetics has based its research on antibodies -- specifically antibodies that can kill cancer cells. Some of the antibodies are engineered to directly kill the cells, similar to the way Genentech's Avastin or ImClone Systems' (Nasdaq: IMCL) Erbitux work. But it is Seattle Genetics' indirect killing mechanism that probably has the best chance of stocking its pipeline.
The company has developed a way to add a toxic drug to an antibody to send the drug specifically to the cancer cells. The beauty of the system is that it can be added to a large number of different antibodies, depending on what types of cells the drug is trying to kill.
Not surprisingly, larger drug companies have become very interested in the system and have licensed Seattle Genetics' technology to develop their own antibody drugs. You can almost hear the cha-ching of royalty payments coming.
But that's not all
The seven compounds in Array's pipeline may look small and immature, but the company doesn't count its partnerships on its main pipeline page. Its phase 2 drug ARRY-886 is partnered with AstraZeneca (NYSE: AZN), and although it showed underwhelming results by itself, the drug may work better in combination with other chemotherapies. Array also has partnered with InterMune on hepatitis C drug ITMN-191, which had pretty good-looking phase 1 clinical trial results.
No such thing as a sure thing
These drug developers are still far from a sure thing. They could run out of money -- or dilute shareholder value by doing many secondary offerings -- and turn out to be lousy investments. But the fact that they all have well-stocked pipelines gives them an edge over other drugmakers.
And if they're able to get just one drug approved, then watch out, because their stock is likely to take off like a rocket. All you have to do is look at the market cap of a one-drug wonder like Onyx Pharmaceuticals or Amylin Pharmaceuticals to realize that these developmental-stage drugmakers have a long way to run if they can get just a single successful drug approved. And that's what makes investing in biotech so much fun.
mfg ipollit
Isis... Isis 353512 ist einer von zwei Kandidaten, die sich in einer klinischen Phase befinden und an denen Isis noch alle Rechte hält
http://www.xconomy.com/san-diego/2008/10/16/isis-pharmaceuti…
Isis Pharmaceuticals’ Second Drug Aims to Block Marker of Heart Disease, Inflammation
Luke Timmerman 10/16/08
Isis Pharmaceuticals CEO Stanley Crooke gets asked a lot about mipomersen, a drug his company is developing to treat dangerously high cholesterol in the blood. It is Isis’s (NASDAQ: ISIS) lead drug candidate and generated a huge partnership with Genzyme worth $325 million in cash upfront, with a lot more to come if this drug pans out in further clinical trials.
Instead of doing the umpteenth interview on that subject, I asked Crooke about other stuff emerging in the Isis pipeline when I stopped by his company’s Carlsbad, CA, offices a couple weeks ago. He looked at me like it was a little odd, but he happily dived in for a discussion about a lesser-known stepchild in the Isis pipeline, called Isis 353512. It’s designed to block a marker of heart disease and other inflammatory conditions, called C-reactive protein.
“This question reminds me a lot of the cholesterol debates 40 years ago,” Crooke says.
Like with those arguments about cholesterol, medical researchers want to know whether the C-reactive protein found in the blood of patients with heart disease is a bad actor that spurs inflammation in vessel walls that can lead to heart attacks, or whether it’s a bystander, Crooke says. The answer has been difficult to pin down because conventional drugs can’t be designed to hit the C-reactive protein. Doctors can already run diagnostic tests of the marker to assess heart disease risk, yet they don’t know whether blocking it can actually lower the risk of heart attacks, or help treat inflammatory diseases like Crohn’s or rheumatoid arthritis, Crooke says. The Isis drug, like all of the company’s candidates in clinical trials, uses proprietary antisense technology to bind specifically with the RNA of the target protein, so it can block the target at its origin in the liver, Crooke says.
The question will be whether that really matters much for patients. Isis is banking on research that shows C-reactive protein is associated with cardiovascular disease patients getting worse, which suggests they may get better if the inflammatory molecule is mopped up. Animal studies of the Isis compound showed it could significantly suppress the protein in the blood and liver.
Isis is currently testing the drug in an early-stage safety study, but it clearly intends to get an answer that will give it confidence to spend money on the kind of bigger trials needed to settle the debate. It is currently testing the drug in a placebo-controlled, randomized study of 58 healthy volunteers to see if it can bring down C-reactive protein levels at a variety of doses. The study is expected to be completed in September 2009—just in time to become fodder for the fall slate of medical meetings next year.
“This is the only drug specific enough to work,” Crooke says. “No other approach has come close to being successful.” He sure makes it sound like he relishes the idea of being first to answer this question. “This is Isis. We lead, not follow.”
mfg ipollit
http://www.xconomy.com/san-diego/2008/10/16/isis-pharmaceuti…
Isis Pharmaceuticals’ Second Drug Aims to Block Marker of Heart Disease, Inflammation
Luke Timmerman 10/16/08
Isis Pharmaceuticals CEO Stanley Crooke gets asked a lot about mipomersen, a drug his company is developing to treat dangerously high cholesterol in the blood. It is Isis’s (NASDAQ: ISIS) lead drug candidate and generated a huge partnership with Genzyme worth $325 million in cash upfront, with a lot more to come if this drug pans out in further clinical trials.
Instead of doing the umpteenth interview on that subject, I asked Crooke about other stuff emerging in the Isis pipeline when I stopped by his company’s Carlsbad, CA, offices a couple weeks ago. He looked at me like it was a little odd, but he happily dived in for a discussion about a lesser-known stepchild in the Isis pipeline, called Isis 353512. It’s designed to block a marker of heart disease and other inflammatory conditions, called C-reactive protein.
“This question reminds me a lot of the cholesterol debates 40 years ago,” Crooke says.
Like with those arguments about cholesterol, medical researchers want to know whether the C-reactive protein found in the blood of patients with heart disease is a bad actor that spurs inflammation in vessel walls that can lead to heart attacks, or whether it’s a bystander, Crooke says. The answer has been difficult to pin down because conventional drugs can’t be designed to hit the C-reactive protein. Doctors can already run diagnostic tests of the marker to assess heart disease risk, yet they don’t know whether blocking it can actually lower the risk of heart attacks, or help treat inflammatory diseases like Crohn’s or rheumatoid arthritis, Crooke says. The Isis drug, like all of the company’s candidates in clinical trials, uses proprietary antisense technology to bind specifically with the RNA of the target protein, so it can block the target at its origin in the liver, Crooke says.
The question will be whether that really matters much for patients. Isis is banking on research that shows C-reactive protein is associated with cardiovascular disease patients getting worse, which suggests they may get better if the inflammatory molecule is mopped up. Animal studies of the Isis compound showed it could significantly suppress the protein in the blood and liver.
Isis is currently testing the drug in an early-stage safety study, but it clearly intends to get an answer that will give it confidence to spend money on the kind of bigger trials needed to settle the debate. It is currently testing the drug in a placebo-controlled, randomized study of 58 healthy volunteers to see if it can bring down C-reactive protein levels at a variety of doses. The study is expected to be completed in September 2009—just in time to become fodder for the fall slate of medical meetings next year.
“This is the only drug specific enough to work,” Crooke says. “No other approach has come close to being successful.” He sure makes it sound like he relishes the idea of being first to answer this question. “This is Isis. We lead, not follow.”
mfg ipollit
Allerdings ist der wichtigste Kandidat in Isis Pipeline Mipomersen, das zusammen mit Genzyme entwickelt wird...
http://www.xconomy.com/san-diego/2008/09/29/genzyme-thinks-s…
Genzyme Thinks Small, and Big, With Cholesterol-Lowering Drug Mipomersen
Luke Timmerman 9/29/08
Genzyme has built an empire by thinking small—treating diseases that affect tiny groups of patients. Now it’s seeking to capitalize on a cholesterol-lowering drug that requires it to think small and big at the same time.
The Cambridge, MA-based biotech company (NASDAQ: GENZ) is starting to show how it intends to make mipomersen into a workhorse for much of its growth over the next decade. I got some insight into the company’s thinking during an interview with John Butler, president of the company’s renal, endocrinology, and cardiovascular business.
Genzyme placed a big bet on mipomersen back in January, when it agreed to pay $325 million upfront to Carlsbad, CA-based Isis Pharmaceuticals (NASDAQ: ISIS) for the right to co-market the drug worldwide. The price for a piece of mipomersen was high for a couple reasons. Soaring cholesterol levels in the fast-food-loving U.S. contribute to heart disease, the nation’s leading cause of death. And drugs that lower cholesterol are the best-selling class of pharmaceutical products, generating 79 million prescriptions a year, according to market research firm IMS Health. Pfizer’s Lipitor, the leader of the class, is the top-selling pharmaceutical ever, with $12.7 billion in sales last year. So anything that comes along that’s more effective, even if it grabs only a sliver of this market, is going to generate serious cash flow.
That’s exactly what Genzyme is banking on. “This offers a significant benefit over the standard of care for a seriously ill patient population. We look at this as a significant driver for us in the future,” Butler says.
A clinical trial last year showed that mipomersen could reduce cholesterol by 40 percent more than statins alone, that it can lower patients’ cholesterol counts to a safe level, and that it can be taken in combination with those drugs. Sounds great, but as Butler says, this isn’t really for everybody. The new drug is only meant to be taken by patients at exceptionally high risk of heart attacks and strokes because of their clogged arteries. Plus, since it’s a weekly injectable, it doesn’t have the same kind of convenience for the masses as a daily pill like Lipitor. (The drug also is designed to work differently than anything on the market. It’s a targeted drug that reduces the production of apoB-100, a protein that’s essential for synthesizing and transporting LDL “bad” cholesterol in the blood.)
“This is not a product that will compete with statins, it’s for patients who aren’t adequately controlled with those drugs,” Butler says.
Mipomersen isn’t scheduled to reach the desk of FDA reviewers until the second half of 2010, so a lot of work still needs to be done to prove it’s ready for the market. Genzyme’s first item on its to-do list is to show that mipomersen works for patients with an extremely rare genetic disorder called homozygous familial hypercholesterolemia that makes it so people can’t properly control cholesterol in the blood. The disorder means that patients have two faulty copies of a gene that controls cholesterol levels, which puts even very young children at risk of a heart attack or stroke, Butler says. About 300 patients, one in a million people in the U.S., have this disorder, and Genzyme expects to be able to ask the FDA for clearance to sell the drug in the second half of 2010 if this trial pans out, Butler says.
That targeting of a small patient population is typical Genzyme. But here’s where the part about thinking bigger enters the picture:
Genzyme’s next step is to show mipomersen can help a broader group of patients—estimated at about 6,000 in the U.S.—who only have one bad copy of the cholesterol-clearing gene, but often have cholesterol scores so high that they need to visit a doctor regularly for what’s called an apheresis blood-filtering procedure. The procedure costs $75,000 to $150,000 a year, Butler says.
Then come the really big numbers. Last month, Genzyme said it started a pivotal trial of the drug for patients with a condition called heterozygous familiar hypercholesterolemia that also have one bad copy of the gene that contributes to their high cholesterol, but it isn’t so severe that they need the blood filtering. About 300,000 patients fall into that category, Butler says. Then there’s another 1 to 2 million patients who are either unable to tolerate statins, or are unable to get their cholesterol low enough by taking those drugs, he says. Two more clinical trials are expected to start later in the year to see if the drug can help those “high-risk” patients, Butler says.
Before Genzyme and Isis can market to that wide of a patient population, Genzyme will need results from what it calls an “outcome” study, Butler says. Genzyme hasn’t yet started the “outcomes” study or talked about its design, but generally the FDA wants to know whether drugs in the category can reduce the death rate and lower the number of heart attacks and strokes, Butler says.
All of this expansion naturally raises the question of price. How do you price a product that’s originally for just 300 seriously ill people (and therefore presumably pretty high), without making it too expensive to reach a mass market?
Butler didn’t want to discuss price with me, not yet anyway. “It’s way too early to talk about price. What drives price is the quality of the data,” he says.
He’s also not talking yet in hard numbers about how much impact mipomersen could have on Genzyme’s future bottom line, but the company has structured its partnership with Isis to prepare for big paydays to come. Genzyme and Isis plan to split profits from the drug, starting with a 70/30 split in favor of Genzyme, with a sliding scale that rises up to a 50/50 split once annual sales reach $2 billion. That might not mean much to a company like Pfizer, which is losing its patent on Lipitor in two years, but it means a lot to Genzyme, which had $3.8 billion in revenue last year as a company.
“This is a product that really fits Genzyme,” Butler says.
mfg ipollit
http://www.xconomy.com/san-diego/2008/09/29/genzyme-thinks-s…
Genzyme Thinks Small, and Big, With Cholesterol-Lowering Drug Mipomersen
Luke Timmerman 9/29/08
Genzyme has built an empire by thinking small—treating diseases that affect tiny groups of patients. Now it’s seeking to capitalize on a cholesterol-lowering drug that requires it to think small and big at the same time.
The Cambridge, MA-based biotech company (NASDAQ: GENZ) is starting to show how it intends to make mipomersen into a workhorse for much of its growth over the next decade. I got some insight into the company’s thinking during an interview with John Butler, president of the company’s renal, endocrinology, and cardiovascular business.
Genzyme placed a big bet on mipomersen back in January, when it agreed to pay $325 million upfront to Carlsbad, CA-based Isis Pharmaceuticals (NASDAQ: ISIS) for the right to co-market the drug worldwide. The price for a piece of mipomersen was high for a couple reasons. Soaring cholesterol levels in the fast-food-loving U.S. contribute to heart disease, the nation’s leading cause of death. And drugs that lower cholesterol are the best-selling class of pharmaceutical products, generating 79 million prescriptions a year, according to market research firm IMS Health. Pfizer’s Lipitor, the leader of the class, is the top-selling pharmaceutical ever, with $12.7 billion in sales last year. So anything that comes along that’s more effective, even if it grabs only a sliver of this market, is going to generate serious cash flow.
That’s exactly what Genzyme is banking on. “This offers a significant benefit over the standard of care for a seriously ill patient population. We look at this as a significant driver for us in the future,” Butler says.
A clinical trial last year showed that mipomersen could reduce cholesterol by 40 percent more than statins alone, that it can lower patients’ cholesterol counts to a safe level, and that it can be taken in combination with those drugs. Sounds great, but as Butler says, this isn’t really for everybody. The new drug is only meant to be taken by patients at exceptionally high risk of heart attacks and strokes because of their clogged arteries. Plus, since it’s a weekly injectable, it doesn’t have the same kind of convenience for the masses as a daily pill like Lipitor. (The drug also is designed to work differently than anything on the market. It’s a targeted drug that reduces the production of apoB-100, a protein that’s essential for synthesizing and transporting LDL “bad” cholesterol in the blood.)
“This is not a product that will compete with statins, it’s for patients who aren’t adequately controlled with those drugs,” Butler says.
Mipomersen isn’t scheduled to reach the desk of FDA reviewers until the second half of 2010, so a lot of work still needs to be done to prove it’s ready for the market. Genzyme’s first item on its to-do list is to show that mipomersen works for patients with an extremely rare genetic disorder called homozygous familial hypercholesterolemia that makes it so people can’t properly control cholesterol in the blood. The disorder means that patients have two faulty copies of a gene that controls cholesterol levels, which puts even very young children at risk of a heart attack or stroke, Butler says. About 300 patients, one in a million people in the U.S., have this disorder, and Genzyme expects to be able to ask the FDA for clearance to sell the drug in the second half of 2010 if this trial pans out, Butler says.
That targeting of a small patient population is typical Genzyme. But here’s where the part about thinking bigger enters the picture:
Genzyme’s next step is to show mipomersen can help a broader group of patients—estimated at about 6,000 in the U.S.—who only have one bad copy of the cholesterol-clearing gene, but often have cholesterol scores so high that they need to visit a doctor regularly for what’s called an apheresis blood-filtering procedure. The procedure costs $75,000 to $150,000 a year, Butler says.
Then come the really big numbers. Last month, Genzyme said it started a pivotal trial of the drug for patients with a condition called heterozygous familiar hypercholesterolemia that also have one bad copy of the gene that contributes to their high cholesterol, but it isn’t so severe that they need the blood filtering. About 300,000 patients fall into that category, Butler says. Then there’s another 1 to 2 million patients who are either unable to tolerate statins, or are unable to get their cholesterol low enough by taking those drugs, he says. Two more clinical trials are expected to start later in the year to see if the drug can help those “high-risk” patients, Butler says.
Before Genzyme and Isis can market to that wide of a patient population, Genzyme will need results from what it calls an “outcome” study, Butler says. Genzyme hasn’t yet started the “outcomes” study or talked about its design, but generally the FDA wants to know whether drugs in the category can reduce the death rate and lower the number of heart attacks and strokes, Butler says.
All of this expansion naturally raises the question of price. How do you price a product that’s originally for just 300 seriously ill people (and therefore presumably pretty high), without making it too expensive to reach a mass market?
Butler didn’t want to discuss price with me, not yet anyway. “It’s way too early to talk about price. What drives price is the quality of the data,” he says.
He’s also not talking yet in hard numbers about how much impact mipomersen could have on Genzyme’s future bottom line, but the company has structured its partnership with Isis to prepare for big paydays to come. Genzyme and Isis plan to split profits from the drug, starting with a 70/30 split in favor of Genzyme, with a sliding scale that rises up to a 50/50 split once annual sales reach $2 billion. That might not mean much to a company like Pfizer, which is losing its patent on Lipitor in two years, but it means a lot to Genzyme, which had $3.8 billion in revenue last year as a company.
“This is a product that really fits Genzyme,” Butler says.
mfg ipollit
Antwort auf Beitrag Nr.: 35.613.315 von ipollit am 18.10.08 14:50:59Ich würde nicht darauf setzten, dass ein EVT-201 auf jeden Fall noch in 2008 zustande kommt.
Nein, falls du meinst, dass ich finanziell auf diesen Zeitraum setze, NÖ mach isch net!
Aber ich denke Evo macht schon Druck, gerade diese Verzögerungen nagen enorm am Kurs, das werden sie hoffentlich wissen. Ich vermute aber, man hat mehrere Optionen geprüft, das hat eben länger gedauert als geplant.
Ich habe es irgendwie im Urin, dass es aber jetzt nicht mehr lange dauert, denke bis Nikolaus ist der Stiefel voll , kann natürlich auch Wunschdenken sein.
VIEL GLÜCK und danke, dass du dich wieder bei Evo "gezeigt" hast, dort haben sie alle den Kopf im Sand, statt die Zeiten zu nutzen, um Schadenswiedergutmachung zu betreiben.
Grüße!
Nein, falls du meinst, dass ich finanziell auf diesen Zeitraum setze, NÖ mach isch net!
Aber ich denke Evo macht schon Druck, gerade diese Verzögerungen nagen enorm am Kurs, das werden sie hoffentlich wissen. Ich vermute aber, man hat mehrere Optionen geprüft, das hat eben länger gedauert als geplant.
Ich habe es irgendwie im Urin, dass es aber jetzt nicht mehr lange dauert, denke bis Nikolaus ist der Stiefel voll
, kann natürlich auch Wunschdenken sein.VIEL GLÜCK und danke, dass du dich wieder bei Evo "gezeigt" hast, dort haben sie alle den Kopf im Sand, statt die Zeiten zu nutzen, um Schadenswiedergutmachung zu betreiben.
Grüße!
ONXX...
http://www.rttnews.com/ArticleView.aspx?Id=771536&pageNum=23…
Onyx Pharma - Awaiting New Beginning
11/11/2008 5:33 AM ET
(RTTNews) - After a strong rally in 2007, shares of Onyx Pharmaceuticals Inc. (ONXX: News ) were hit hard in 2008 and now trade around $30, roughly half their year's high. After all, being in a dicey business where fortunes rise or fall based on the success of a single drug, the stock price reversal of Onyx is no surprise.
But with the company's key cancer drug Nexavar having blockbuster potential, Onyx may find itself back in the spotlight. Nexavar is developed by Onyx and Bayer HealthCare Pharmaceuticals Inc. Onyx funds 50% of the development costs for Nexavar worldwide, except in Japan. Everywhere else in the world, except in Japan, the two companies equally split the profits or losses of the drug. In Japan, Bayer funds all development cost for Nexavar, and Onyx receives a high single-digit royalty on sales.
February Blues - Nexavar Fails In Lung Cancer Trial
Closing at $55.62, the stock ended 2007 with its biggest-ever annual percentage gain of 418%. But Nexavar's failure in a late-stage trial for lung cancer in early February of this year thwarted the stock's upward momentum. However, continued growth of Nexavar has piqued investor's interest, resulting in a rebound in the company's stock price.
In late February, following Nexavar's failure to meet its primary goal of improving overall survival, Onyx and Bayer halted a phase III study testing Nexavar against non-small cell lung cancer, or NSCLC when administered in combination with chemotherapeutic agents carboplatin and paclitaxel.
In the phase III study dubbed ESCAPE (Evaluation of Sorafenib, Carboplatin And Paclitaxel Efficacy in NSCLC), higher mortality was observed in the subset of patients with squamous cell carcinoma of the lung treated with Nexavar in combination with two types of chemotherapeutic agents, compared to those treated with the chemotherapeutic agents alone.
This is not the first time that Onyx is pulling the plug on a late-stage trial of Nexavar. In December 2006, the company halted a phase III trial, which was designed to test the effectiveness of Nexavar in combination with chemotherapeutic agents against melanoma, the most serious type of skin cancer. The trial failed to meet its primary endpoint of progression-free survival, which is defined as the time that a patient lives without meaningful tumor growth.
Despite the failures, Onyx remains committed in maximizing the potential of Nexavar. In 2005, Nexavar was approved by the FDA for treating advanced forms of kidney cancer. The drug won the FDA approval in November 2007 for expanded use in the treatment of a form of liver cancer known as hepatocellular carcinoma, when the cancer is inoperable. Nexavar is currently being investigated in several ongoing trials in non-small cell lung cancer, melanoma, breast cancer and other tumor types.
Impressive Nexavar Sales Growth
In the United States, Nexavar competes with Pfizer Inc.'s (PFE) Sutent and Wyeth's (WYE) Torisel. Nexavar continues to gain widespread acceptance across the globe, spurring the drug's sales. The net proceeds from the drug's sales after costs are shared equally between Onyx and Bayer worldwide, except in Japan.
In January, the drug was approved in Japan for the treatment of renal cell carcinoma, a form of kidney cancer. The drug is the first oral targeted therapy to be approved in Japan. Renal cell carcinoma is the most common form of kidney cancer and accounts for around 8,000 new cases per year in Japan alone.
In July, Nexavar was approved in China for the treatment of liver cancer. In the Asia-Pacific region, over 8% of the general population is infected with chronic hepatitis B, while 2%-4% is infected with chronic hepatitis C. Both infections are the leading causes of primary liver cancer worldwide. Nexavar has also been approved for liver cancer in South Korea.
In Japan, the application seeking approval of Nexavar for liver cancer is pending and the drug is expected to be launched in the first half of 2009. Annual deaths due to liver cancer in Japan are estimated at over 35,000. Expanding the drug's label will further boost Nexavar's sales in Japan. In Japan, Onyx gets royalties on sales of Nexavar.
Despite heightened competition, Nexavar raked in sales of $181 million in the third quarter ended September 30, 2008, an increase of an impressive 73% over the comparable quarter last year. According to the company, approximately $133 million of the sales were generated outside the United States and about $48 million were generated within the United States.
In the European Union, Nexavar has been launched for liver cancer in Germany, France, Spain, Italy and Greece. The drug is currently approved in more than 60 countries for the treatment of liver cancer and in over 70 countries for treating kidney cancer. According to the company, most launches worldwide in the kidney cancer market have now been completed.
The ongoing global launches of Nexavar and successful outreach to new patient population, bodes well for Onyx.
Solid Showing; Yet Conservative Outlook
Onyx Pharma is headed by Tony Coles who took over as CEO in July. On the first-quarter earnings conference call Tony Coles said that he expects the company to be "breakeven or profitable on the bottom line for the full year 2008".
On a GAAP basis, Onyx's net income for the third-quarter ended September 30, 2008, surged up to $12.2 million or $0.21 per share from $0.6 million or $0.01 per share in the same period in 2007.
Excluding employee stock-based compensation expense, non-GAAP net income for the quarter rose nearly four-fold to $16.6 million or $0.29 per share from $4.2 million or $0.08 per share in the year-ago quarter. Wall Street analysts were expecting the company to earn $0.02 per share.
Quarterly net revenue from unconsolidated joint business was $39.9 million compared to $17.6 million for the same period in 2007.
For the full year 2008, analysts have revised their projections within the past 7 days, raising the consensus earnings estimate by a penny to $0.34 per share.
Onyx has never provided sales guidance in its history before. But that has changed under the tenure of Coles who is willing to provide as much clarity as possible to everyone on Wall Street.
In May, the company said that it expects Nexavar sales for 2008 to range between $600 million and $650 million. Nexavar net sales for the nine months ended September 30, 2008 were $501.3 million, compared to $246.8 million in the year-ago period.
Onyx, which has long remained a one-trick pony dependant on Nexavar, licensed an anticancer compound, BGC 945 from London-based BTG plc. in a deal valued at $320 million early this month. That seems to be a wise move on the company's part to build its pipeline and moderate any risk associated with being a one-product business. BGC 945, which has been renamed as ONX 0801 by Onyx, is currently in late-stage preclinical development.
Under the terms of the licensing deal, BTG will receive $13 million upfront and has the potential to receive development milestone payments of up to $72 million plus additional payments of up to $235 million for product approval and commercialization milestones. BTG will also receive a royalty on any future sales worldwide.
One of the lead compounds - PD 332991, a cyclin-dependent kinase inhibitor, identified by Onyx under a research collaboration with Warner-Lambert Co., a subsidiary of Pfizer, entered clinical testing in September 2004. Upon the commercialization of PD 332991 by Pfizer, Onyx will receive milestone payments and royalties on worldwide sales.
Conclusion
Onyx will continue to ride on Nexavar's success, and approval in additional indications will further boost the drug's sales. That said, the company could see downside if it fails to obtain label expansion for Nexavar, which exemplifies the risk-fraught nature of the biotech business.
by RTT Staff Writer
mfg ipollit
http://www.rttnews.com/ArticleView.aspx?Id=771536&pageNum=23…
Onyx Pharma - Awaiting New Beginning
11/11/2008 5:33 AM ET
(RTTNews) - After a strong rally in 2007, shares of Onyx Pharmaceuticals Inc. (ONXX: News ) were hit hard in 2008 and now trade around $30, roughly half their year's high. After all, being in a dicey business where fortunes rise or fall based on the success of a single drug, the stock price reversal of Onyx is no surprise.
But with the company's key cancer drug Nexavar having blockbuster potential, Onyx may find itself back in the spotlight. Nexavar is developed by Onyx and Bayer HealthCare Pharmaceuticals Inc. Onyx funds 50% of the development costs for Nexavar worldwide, except in Japan. Everywhere else in the world, except in Japan, the two companies equally split the profits or losses of the drug. In Japan, Bayer funds all development cost for Nexavar, and Onyx receives a high single-digit royalty on sales.
February Blues - Nexavar Fails In Lung Cancer Trial
Closing at $55.62, the stock ended 2007 with its biggest-ever annual percentage gain of 418%. But Nexavar's failure in a late-stage trial for lung cancer in early February of this year thwarted the stock's upward momentum. However, continued growth of Nexavar has piqued investor's interest, resulting in a rebound in the company's stock price.
In late February, following Nexavar's failure to meet its primary goal of improving overall survival, Onyx and Bayer halted a phase III study testing Nexavar against non-small cell lung cancer, or NSCLC when administered in combination with chemotherapeutic agents carboplatin and paclitaxel.
In the phase III study dubbed ESCAPE (Evaluation of Sorafenib, Carboplatin And Paclitaxel Efficacy in NSCLC), higher mortality was observed in the subset of patients with squamous cell carcinoma of the lung treated with Nexavar in combination with two types of chemotherapeutic agents, compared to those treated with the chemotherapeutic agents alone.
This is not the first time that Onyx is pulling the plug on a late-stage trial of Nexavar. In December 2006, the company halted a phase III trial, which was designed to test the effectiveness of Nexavar in combination with chemotherapeutic agents against melanoma, the most serious type of skin cancer. The trial failed to meet its primary endpoint of progression-free survival, which is defined as the time that a patient lives without meaningful tumor growth.
Despite the failures, Onyx remains committed in maximizing the potential of Nexavar. In 2005, Nexavar was approved by the FDA for treating advanced forms of kidney cancer. The drug won the FDA approval in November 2007 for expanded use in the treatment of a form of liver cancer known as hepatocellular carcinoma, when the cancer is inoperable. Nexavar is currently being investigated in several ongoing trials in non-small cell lung cancer, melanoma, breast cancer and other tumor types.
Impressive Nexavar Sales Growth
In the United States, Nexavar competes with Pfizer Inc.'s (PFE) Sutent and Wyeth's (WYE) Torisel. Nexavar continues to gain widespread acceptance across the globe, spurring the drug's sales. The net proceeds from the drug's sales after costs are shared equally between Onyx and Bayer worldwide, except in Japan.
In January, the drug was approved in Japan for the treatment of renal cell carcinoma, a form of kidney cancer. The drug is the first oral targeted therapy to be approved in Japan. Renal cell carcinoma is the most common form of kidney cancer and accounts for around 8,000 new cases per year in Japan alone.
In July, Nexavar was approved in China for the treatment of liver cancer. In the Asia-Pacific region, over 8% of the general population is infected with chronic hepatitis B, while 2%-4% is infected with chronic hepatitis C. Both infections are the leading causes of primary liver cancer worldwide. Nexavar has also been approved for liver cancer in South Korea.
In Japan, the application seeking approval of Nexavar for liver cancer is pending and the drug is expected to be launched in the first half of 2009. Annual deaths due to liver cancer in Japan are estimated at over 35,000. Expanding the drug's label will further boost Nexavar's sales in Japan. In Japan, Onyx gets royalties on sales of Nexavar.
Despite heightened competition, Nexavar raked in sales of $181 million in the third quarter ended September 30, 2008, an increase of an impressive 73% over the comparable quarter last year. According to the company, approximately $133 million of the sales were generated outside the United States and about $48 million were generated within the United States.
In the European Union, Nexavar has been launched for liver cancer in Germany, France, Spain, Italy and Greece. The drug is currently approved in more than 60 countries for the treatment of liver cancer and in over 70 countries for treating kidney cancer. According to the company, most launches worldwide in the kidney cancer market have now been completed.
The ongoing global launches of Nexavar and successful outreach to new patient population, bodes well for Onyx.
Solid Showing; Yet Conservative Outlook
Onyx Pharma is headed by Tony Coles who took over as CEO in July. On the first-quarter earnings conference call Tony Coles said that he expects the company to be "breakeven or profitable on the bottom line for the full year 2008".
On a GAAP basis, Onyx's net income for the third-quarter ended September 30, 2008, surged up to $12.2 million or $0.21 per share from $0.6 million or $0.01 per share in the same period in 2007.
Excluding employee stock-based compensation expense, non-GAAP net income for the quarter rose nearly four-fold to $16.6 million or $0.29 per share from $4.2 million or $0.08 per share in the year-ago quarter. Wall Street analysts were expecting the company to earn $0.02 per share.
Quarterly net revenue from unconsolidated joint business was $39.9 million compared to $17.6 million for the same period in 2007.
For the full year 2008, analysts have revised their projections within the past 7 days, raising the consensus earnings estimate by a penny to $0.34 per share.
Onyx has never provided sales guidance in its history before. But that has changed under the tenure of Coles who is willing to provide as much clarity as possible to everyone on Wall Street.
In May, the company said that it expects Nexavar sales for 2008 to range between $600 million and $650 million. Nexavar net sales for the nine months ended September 30, 2008 were $501.3 million, compared to $246.8 million in the year-ago period.
Onyx, which has long remained a one-trick pony dependant on Nexavar, licensed an anticancer compound, BGC 945 from London-based BTG plc. in a deal valued at $320 million early this month. That seems to be a wise move on the company's part to build its pipeline and moderate any risk associated with being a one-product business. BGC 945, which has been renamed as ONX 0801 by Onyx, is currently in late-stage preclinical development.
Under the terms of the licensing deal, BTG will receive $13 million upfront and has the potential to receive development milestone payments of up to $72 million plus additional payments of up to $235 million for product approval and commercialization milestones. BTG will also receive a royalty on any future sales worldwide.
One of the lead compounds - PD 332991, a cyclin-dependent kinase inhibitor, identified by Onyx under a research collaboration with Warner-Lambert Co., a subsidiary of Pfizer, entered clinical testing in September 2004. Upon the commercialization of PD 332991 by Pfizer, Onyx will receive milestone payments and royalties on worldwide sales.
Conclusion
Onyx will continue to ride on Nexavar's success, and approval in additional indications will further boost the drug's sales. That said, the company could see downside if it fails to obtain label expansion for Nexavar, which exemplifies the risk-fraught nature of the biotech business.
by RTT Staff Writer
mfg ipollit
Änderungen im Depot: Intercell und OSIP herausgenommen, Rigel etwas aufgestockt, neue Position Medigene
mfg ipollit
mfg ipollit
Genmab...
weiter positive Daten zu Arzerra (Ofatumumab) in CLL. Antrag auf Zulassung sollte bald gestellt werden.
Merrill goes to BUY from Neutral, target 335
Strong safety and efficacy support approval in CLL
We attended the oral data presentation of Genmab’s ofatumumab phase III trial in relapsed/refractory CLL (a blood cancer) at the ASH clinical meeting. The data, described as "quite stunning" by a leading CLL clinician, showed impressive response rates across all patient subgroups, including patients previously exposed to Rituxan, another antibody commonly used to treat CLL. Clinicians also thought ofatumumab was safe and well tolerated. In our view the data strongly support approval of ofatumumab in relapsed/refractory CLL, for which Genmab and partner GSK plan to file in January 2009. We have increased our CLL peak sales forecasts from $350mn to $500mn and upgrade to Buy from Neutral.
High response rates across patient subgroups
Response rates were consistently high in all patient subgroup analyses performed (47-50+%), including prior exposure to the Rituxan. 54% of double refractory (DR) patients previously exposed to Rituxan achieved a response vs. 63% of those not exposed. The response rates in the Campath-ineligible (BFR) group were 44%
and 50%. It is not clear whether ofatumumab is effective in Rituxan refractory patients, however, as it is difficult to discern whether patients were refractory to the chemotherapy or to Rituxan itself.
Safety profile benign, impressive survival
Clinicians we spoke to had no concerns about ofatumumab's safety profile, and most thought it was comparable to Rituxan's. The 25% infection rate was not seen as an issue, as late-stage CLL patients are generally immunosuppressed and hence highly susceptible to infections regardless of therapy. Clinicians were both
surprised and impressed by the high response rates achieved. Historical analysis and their own data demonstrate that these patients do not respond to antibody therapy and survive for only 9-10 months (vs. 14-15 months in this study).
Duration of response slightly disappointing to some
Patients who responded to ofatumumab maintained their response for 6-7 months and experienced progression-free survival (PFS) of 5.7 (DR) and 5.9 (BFR) months. While this is well above the 3-4 months indicated by FDA for approval, some clinicians voiced disappointment, particularly given the strength of the response rates. However, it needs to be taken into account that the study enrolled
a very challenging patient population.
Upgrading to Buy from Neutral; PO unchanged at DKK325
Both the quality of the clinical data presented at ASH and clinicians’ reception of the data exceeded our expectations. Even though we see some potential longer term risks regarding commercial success in other indications (eg rheumatoid arthritis), we expect the shares to perform well over the near term.
mfg ipollit
weiter positive Daten zu Arzerra (Ofatumumab) in CLL. Antrag auf Zulassung sollte bald gestellt werden.
Merrill goes to BUY from Neutral, target 335
Strong safety and efficacy support approval in CLL
We attended the oral data presentation of Genmab’s ofatumumab phase III trial in relapsed/refractory CLL (a blood cancer) at the ASH clinical meeting. The data, described as "quite stunning" by a leading CLL clinician, showed impressive response rates across all patient subgroups, including patients previously exposed to Rituxan, another antibody commonly used to treat CLL. Clinicians also thought ofatumumab was safe and well tolerated. In our view the data strongly support approval of ofatumumab in relapsed/refractory CLL, for which Genmab and partner GSK plan to file in January 2009. We have increased our CLL peak sales forecasts from $350mn to $500mn and upgrade to Buy from Neutral.
High response rates across patient subgroups
Response rates were consistently high in all patient subgroup analyses performed (47-50+%), including prior exposure to the Rituxan. 54% of double refractory (DR) patients previously exposed to Rituxan achieved a response vs. 63% of those not exposed. The response rates in the Campath-ineligible (BFR) group were 44%
and 50%. It is not clear whether ofatumumab is effective in Rituxan refractory patients, however, as it is difficult to discern whether patients were refractory to the chemotherapy or to Rituxan itself.
Safety profile benign, impressive survival
Clinicians we spoke to had no concerns about ofatumumab's safety profile, and most thought it was comparable to Rituxan's. The 25% infection rate was not seen as an issue, as late-stage CLL patients are generally immunosuppressed and hence highly susceptible to infections regardless of therapy. Clinicians were both
surprised and impressed by the high response rates achieved. Historical analysis and their own data demonstrate that these patients do not respond to antibody therapy and survive for only 9-10 months (vs. 14-15 months in this study).
Duration of response slightly disappointing to some
Patients who responded to ofatumumab maintained their response for 6-7 months and experienced progression-free survival (PFS) of 5.7 (DR) and 5.9 (BFR) months. While this is well above the 3-4 months indicated by FDA for approval, some clinicians voiced disappointment, particularly given the strength of the response rates. However, it needs to be taken into account that the study enrolled
a very challenging patient population.
Upgrading to Buy from Neutral; PO unchanged at DKK325
Both the quality of the clinical data presented at ASH and clinicians’ reception of the data exceeded our expectations. Even though we see some potential longer term risks regarding commercial success in other indications (eg rheumatoid arthritis), we expect the shares to perform well over the near term.
mfg ipollit
Onyx... Nexavar bietet weiter Potential
Onyx Pharma upgraded
Thursday December 11, 8:41 am ET
Onyx upgraded as analyst expects wider Nexavar use and greater treatment time in Europe
NEW YORK (AP) -- A JPMorgan analyst upgraded shares of Onyx Pharmaceuticals Inc. Thursday, predicting increased use of the company's liver cancer drug Nexavar in Europe.
Cory Kasimov upgraded the stock to "Overweight" from "Neutral" based on a JPMorgan survey of 50 European doctors. He said the results show that Nexavar use against both early stage and advanced liver cancer will increase, and doctors are using the drug for longer periods of time as they grow more familiar with it.
He added that many physicians are willing to prescribe Nexavar as an adjuvant therapy, which aims to prevent the disease form returning after it has been treated with surgery or radiation, if clinical trial results show Nexavar is effective in that setting.
Under a licensing agreement, Bayer Healthcare Pharmaceuticals receives all the revenue from Nexavar sales, and Onyx gets milestone payments.
Within a year, Nexavar will be used in 61 percent of advanced liver cancer cases and 34 percent of early stage cases, Kasimov said. That compares to 49 percent and 18 percent currently, in his estimation. He said the survey also shows doctors in Germany, where the drug was first approved, are prescribing it for longer periods of time, and physicians in other countries may do the same.
The analyst said Onyx shares are attractive because the company should post revenue growth of 60 percent per year through 2012, and Onyx is not carrying any debt.
Last Thursday, Cowen & Co. analyst Phil Nadeau said he expects Onyx's stock to outperform the market by 20 percent over the next 12 months as investors gain confidence in Nexavar. Nadeau sees Asia as a key market for the company but said hurdles include the drug's price and lack of reimbursement.
Shares of Onyx closed at $30.09 on Wednesday. While the stock has climbed 24 percent since Nov. 21, that is barely half what the stock was worth in August.
mfg ipollit
Onyx Pharma upgraded
Thursday December 11, 8:41 am ET
Onyx upgraded as analyst expects wider Nexavar use and greater treatment time in Europe
NEW YORK (AP) -- A JPMorgan analyst upgraded shares of Onyx Pharmaceuticals Inc. Thursday, predicting increased use of the company's liver cancer drug Nexavar in Europe.
Cory Kasimov upgraded the stock to "Overweight" from "Neutral" based on a JPMorgan survey of 50 European doctors. He said the results show that Nexavar use against both early stage and advanced liver cancer will increase, and doctors are using the drug for longer periods of time as they grow more familiar with it.
He added that many physicians are willing to prescribe Nexavar as an adjuvant therapy, which aims to prevent the disease form returning after it has been treated with surgery or radiation, if clinical trial results show Nexavar is effective in that setting.
Under a licensing agreement, Bayer Healthcare Pharmaceuticals receives all the revenue from Nexavar sales, and Onyx gets milestone payments.
Within a year, Nexavar will be used in 61 percent of advanced liver cancer cases and 34 percent of early stage cases, Kasimov said. That compares to 49 percent and 18 percent currently, in his estimation. He said the survey also shows doctors in Germany, where the drug was first approved, are prescribing it for longer periods of time, and physicians in other countries may do the same.
The analyst said Onyx shares are attractive because the company should post revenue growth of 60 percent per year through 2012, and Onyx is not carrying any debt.
Last Thursday, Cowen & Co. analyst Phil Nadeau said he expects Onyx's stock to outperform the market by 20 percent over the next 12 months as investors gain confidence in Nexavar. Nadeau sees Asia as a key market for the company but said hurdles include the drug's price and lack of reimbursement.
Shares of Onyx closed at $30.09 on Wednesday. While the stock has climbed 24 percent since Nov. 21, that is barely half what the stock was worth in August.
mfg ipollit
Exelixis... funktioniert das Geschäftsmodell doch? GSK hat zuvor die Option für XL184 nicht wahrgenommen...
EXEL hat nun doch einen Partner gefundnen: BMS!... 240 Mio USD Upfront für eine gemeinsame Entwicklung von XL184 und XL281 sind schon eine gewaltige Summe!
240 Mio USD... die aktuelle MK von EXEL beträgt dagegen 400 Mio USD mit hohem Cashburn...
Biotech Bristol-Myers, Exelixis in Drug Pact: Report
12/12/08 - 01:26 AM EST
The agreement, to be announced Friday, has been reached amid turmoil in the biotechnology industry, where the financial crisis has shut hundreds of companies out of the capital markets, the Wall Street Journal reports.
The Journal cites information from industry trade group BIO which says that 180, or 45%, of the 370 publicly traded biotech companies have less than one year of cash on hand, and most have little or no revenue.
Under the agreement, Bristol-Myers will pay Exelixis $195 million now and another $45 million in 2009 to co-develop the drugs, known as XL184 and XL281. The Journal reports several hundred million dollars in additional payments would follow if the drugs pass development and regulatory milestones and hit certain sales targets.
The $240 million upfront payment is one of the biggest amounts large pharmaceutical companies have paid in similar agreements, the newspaper reports.
Testing for XL184 is most advanced for thyroid cancer, but it is also in human studies for a brain cancer known as glioblastoma and for lung cancer. XL281 is in early-stage testing for several solid tumors, according to the Journal.
mfg ipollit
EXEL hat nun doch einen Partner gefundnen: BMS!... 240 Mio USD Upfront für eine gemeinsame Entwicklung von XL184 und XL281 sind schon eine gewaltige Summe!
240 Mio USD... die aktuelle MK von EXEL beträgt dagegen 400 Mio USD mit hohem Cashburn...
Biotech Bristol-Myers, Exelixis in Drug Pact: Report
12/12/08 - 01:26 AM EST
The agreement, to be announced Friday, has been reached amid turmoil in the biotechnology industry, where the financial crisis has shut hundreds of companies out of the capital markets, the Wall Street Journal reports.
The Journal cites information from industry trade group BIO which says that 180, or 45%, of the 370 publicly traded biotech companies have less than one year of cash on hand, and most have little or no revenue.
Under the agreement, Bristol-Myers will pay Exelixis $195 million now and another $45 million in 2009 to co-develop the drugs, known as XL184 and XL281. The Journal reports several hundred million dollars in additional payments would follow if the drugs pass development and regulatory milestones and hit certain sales targets.
The $240 million upfront payment is one of the biggest amounts large pharmaceutical companies have paid in similar agreements, the newspaper reports.
Testing for XL184 is most advanced for thyroid cancer, but it is also in human studies for a brain cancer known as glioblastoma and for lung cancer. XL281 is in early-stage testing for several solid tumors, according to the Journal.
mfg ipollit
Beitrag zu dieser Diskussion schreiben
Zu dieser Diskussion können keine Beiträge mehr verfasst werden, da der letzte Beitrag vor mehr als zwei Jahren verfasst wurde und die Diskussion daraufhin archiviert wurde.
Bitte wenden Sie sich an feedback@wallstreet-online.de und erfragen Sie die Reaktivierung der Diskussion oder starten Sie eine neue Diskussion.
Meistdiskutiert
| Wertpapier | Beiträge | |
|---|---|---|
| 204 | ||
| 93 | ||
| 89 | ||
| 66 | ||
| 65 | ||
| 57 | ||
| 42 | ||
| 39 | ||
| 36 | ||
| 28 |
| Wertpapier | Beiträge | |
|---|---|---|
| 25 | ||
| 25 | ||
| 22 | ||
| 21 | ||
| 20 | ||
| 20 | ||
| 20 | ||
| 17 | ||
| 17 | ||
| 15 |



















